Data dredging or a $50B drug: an intro to interpreting clinical data
by Richard Murphey
In this post, we'll introduce basic concepts in interpreting clinical data using a real-world example: a case study of Biogen's aducanumab. Aducanumab is being developed as a treatment for Alzheimer's disease, and could be the first disease-modifying therapy for one of the largest and fastest growing causes of death in the US.
But does it work? That is one of the biggest questions in the biotech world today.
In this post, I'll provide a bit of background on the aducanumab situation. Then we will walk through the slides that Biogen used to announce what STAT News calls “the most shocking biotech news in all of 2019”: Biogen's decision to submit aducanumab for approval, just months after announcing it was canceling the program after a failed pivotal study (STAT’s podcast on this topic is well worth a listen). I'll walk through my interpretation of the data, and hopefully will provide enough context for you to draw your own conclusions.
I'll note that this is intended to be an introduction to analyzing clinical trials through a real life analysis. It isn't meant to be a definitive or expert analysis, nor is it an investment recommendation. I have no position in Biogen or intention of initiating a position in the near future.
Background on aducanumab
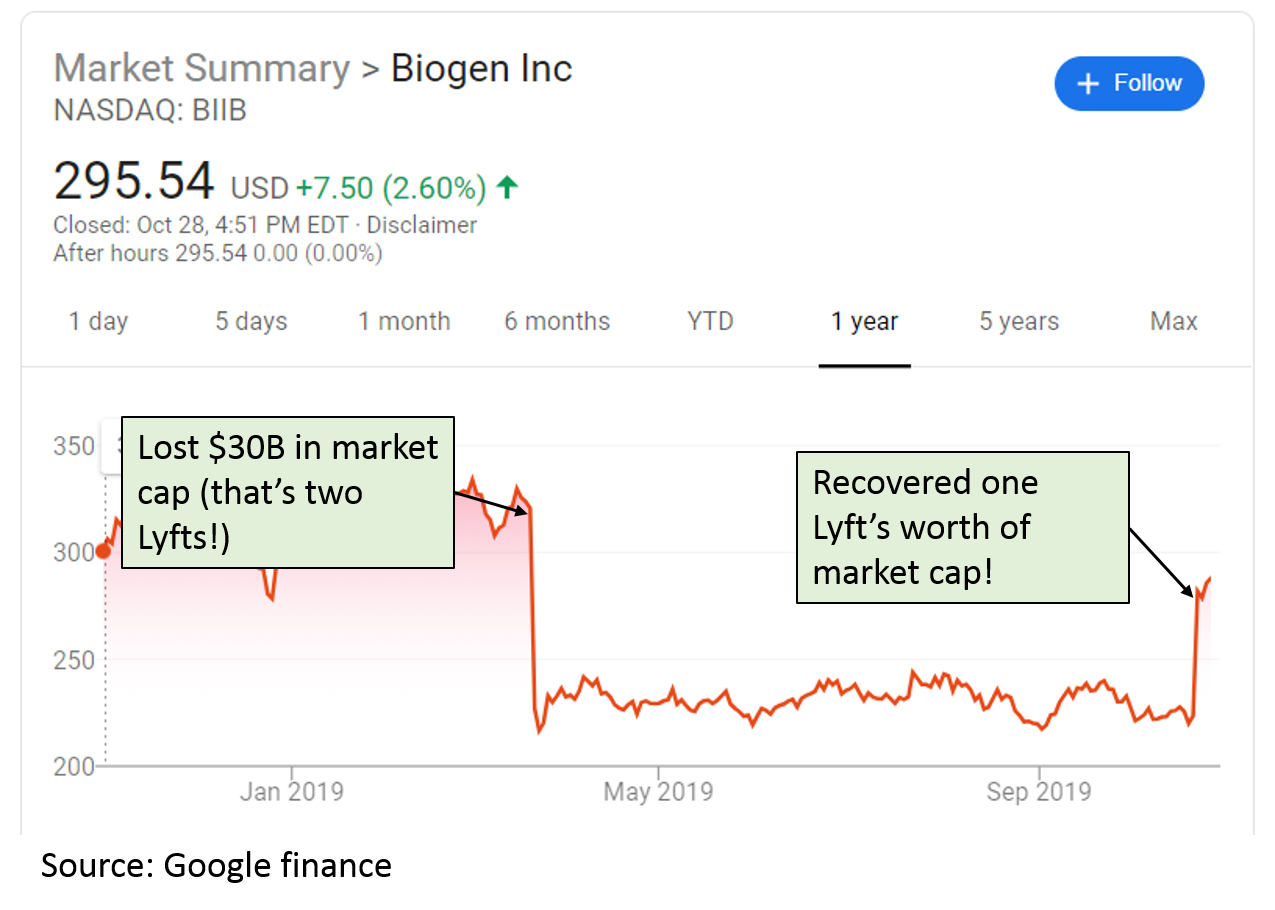
In October 2019, Biogen announced that it was submitting its potential Alzheimer’s drug, aducanumab, for FDA approval. This was shocking because a few months prior, Biogen announced it was stopping development of this drug because one of its pivotal trials failed -- their drug did no better than placebo at improving Alzheimer's symptoms like dementia. Because aducanumab is potentially the first disease-modifying treatment for Alzheimer's, it could be one of the best-selling drugs of all time. Biogen's stock lost $30B of market value when it announced the study failure.
In its recent announcement, Biogen said that it had re-analyzed the data from the "futility analysis" that led to the study being declared a failure, and that if you cut the data a certain way, the study shows the drug might work. Biogen said there was a protocol amendment -- a change to the design of a study -- that rendered the futility analysis invalid. Biogen gained $15B in market cap on the news.
The big question is: does this drug actually work, or is Biogen just data dredging? And another big question is will FDA approve the drug, even if it isn’t clear that it helps patients with Alzheimer’s?
To evaluate whether the drug works, let’s look at the data. Check out the slides Biogen’s press release announcing that they decided to submit the drug as our primary source.
Quick background on FDA submission
Before we get into the slides, some context.
The news here is that Biogen decided to “submit a regulatory filing” for aducanumab. What does it mean to “submit a regulatory filing”, and why is this important?
In order to sell a drug to treat disease in the US, companies have to get FDA approval. Companies must conduct many studies to show that the drug is safe and effective. If the company believes the drug to be safe and effective, it can decide to submit an application to FDA. This application consists of an enormous amount of data from the studies of the drug and must be prepared in accordance with FDA guidelines. FDA will review this data and decide whether to approve it.
Typically, in order to get FDA approval, companies must demonstrate that their drug is safe and effective in two large, well-controlled studies (or in some cases, one well-controlled study and "confirmatory evidence"). Biogen conducted two such studies of its drug aducanumab: the EMERGE study and the ENGAGE study.
The data from the EMERGE study was promising. But in March 2019, Biogen announced that the ENGAGE study failed. Thus Biogen did not have sufficient data to support an FDA approval: it had one good Phase 3 study and one failed Phase 3 study.
Biogen's recent announcement is that if you look at the data a certain way, ENGAGE is positive for a subset of patients. So for regulatory purposes, EMERGE is "one well-controlled study" and ENGAGE could qualify as "confirmatory evidence". Whether FDA will agree with that is unclear. It is also unclear whether FDA will be ok with one good study + confirmatory evidence for Alzheimer's drugs, or if they will require the more stringent standard of two well-controlled studies for approval.
One more piece of context: aducanumab is designed to treat Alzheimer’s by reducing the buildup of a protein called beta amyloid in neurons. The “beta amyloid” hypothesis states that buildup of toxic beta amyloid causes Alzheimer’s. According to this hypothesis, drugs that reduce the buildup of beta amyloid should slow progression of Alzheimer’s.
Until the last few years, this has been the dominant therapeutic hypothesis for Alzheimer’s. But pretty much every large study of drugs targeting beta amyloid have failed – until Biogen’s EMERGE study. The subsequent failure of ENGAGE was thought to be the nail in the coffin for this theory.
With that context in hand, let's look at Biogen's slides.
The clinical data slides
First, I’ll discuss the two pivotal clinical studies Biogen conducted to test whether aducanumab is safe and effective in treating Alzheimer’s. Then I’ll discuss Biogen’s recent re-analysis.
We'll start with slide 6 in Biogen's presentation.
Slide 6: study design
Before analyzing the results of a study, it is important to understand the design: what were the characteristics of patients who were enrolled, what drug was being tested and at what doses, what were the controls, how many patients were enrolled, was the study randomized and blinded?
It is also important to know what endpoints are being measured. The "primary endpoint" generally tests the main hypothesis of a study and is the single largest determinant of whether a study is viewed as "successful". In large Phase 3 studies like this, the primary endpoint is usually a measure of whether important symptoms or other clinical outcomes (like survival, rate of heart attacks, etc.) improved during the course of a study. Investigators can compare whether patients receiving the drug improved on the primary endpoint more than did the patients receiving placebo. If the improvement is higher in the drug arm, and that improvement is statistically significant according to a predefined statistical analysis plan, then the study supports the hypothesis that the drug works.
Investigators also look at secondary endpoints. These are often other measures of safety and effectiveness that can support or refute the primary endpoint. These can also enable exploratory analyses that help researchers develop hypotheses to test in future studies.
We won't go into too much detail on the study design in this post. We'll focus on the slide presented here.
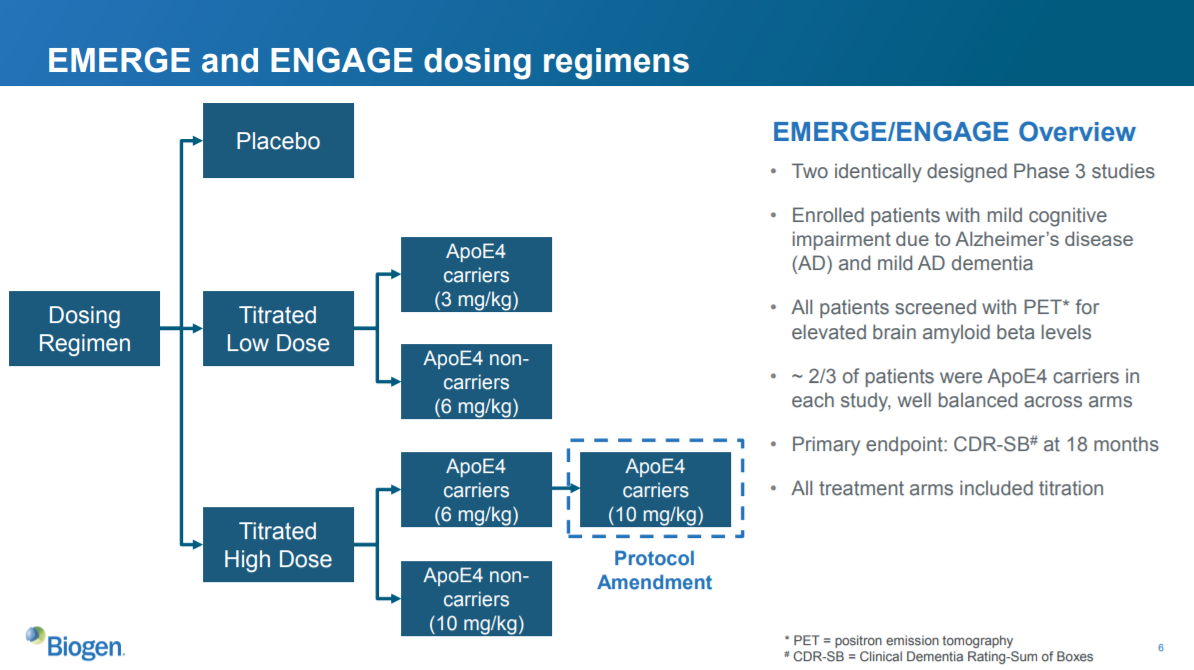
Biogen's study design
This slide is a high-level description of the study design of EMERGE and ENGAGE. These studies tested whether Biogen’s drug, aducanumab, was effective in treating Alzheimer’s compared to placebo. The “primary endpoint” used to measure progression of Alzheimer’s was CDR-SB at 18 months. CDR-SB is a scale that measures dementia. They also looked at other endpoints measuring different aspects of AD, and they looked at safety of the drug.
Both of these studies had identical designs, with one minor exception that was caused by the protocol amendment (we'll discuss this later):
- They enrolled patients with mild cognitive impairment and mild AD dementia. Prior studies suggest that it is challenging to slow decline in patients with more significant cognitive impairment, but drugs could potentially help more mild patients.
- They screened all patients to make sure they had elevated levels of amyloid beta in the brain. Remember, this drug targets amyloid beta; too much amyloid beta is believed to cause AD. Prior studies of anti-amyloid beta drugs didn’t always include this check, and some believe this was a reason other studies failed.
- ApoE4 is a genetic mutation associated with higher risk of getting AD, and getting AD at a younger age. If one arm of the study had more ApoE4 patients, those patients could decline at a different rate than non-ApoE4 patients, independent of whether the treatment is working. This would confound the results of the study because you wouldn’t be able to tell if patients in the high-ApoE4 arm are doing worse because of the drug they get, or because their disease progresses differently.
- The primary endpoint is CDR-SB at 18 months. CDR-SB is a validated scale used to measure dementia levels. So the study is measuring whether the drug slows progression of dementia after 18 months. It is essential to specifically define the primary endpoint, as well as the statistical plan for analyzing the endpoint, before starting the study. More on this later.
The three arms of the studies were:
- Placebo
- Low dose
- High dose
The low dose and high dose arms were subdivided into ApoE4 carriers and non-carriers (see the third bullet point above). ApoE4 carriers got a lower dose than non-carriers (I don’t know why). The protocol amendment increased the high dose for ApoE4 carriers from 6 mg/kg to 10 mg/kg.
Did these studies show that aducanumab was an effective treatment for AD? Let’s look at the data.
Slide 10: EMERGE primary endpoint
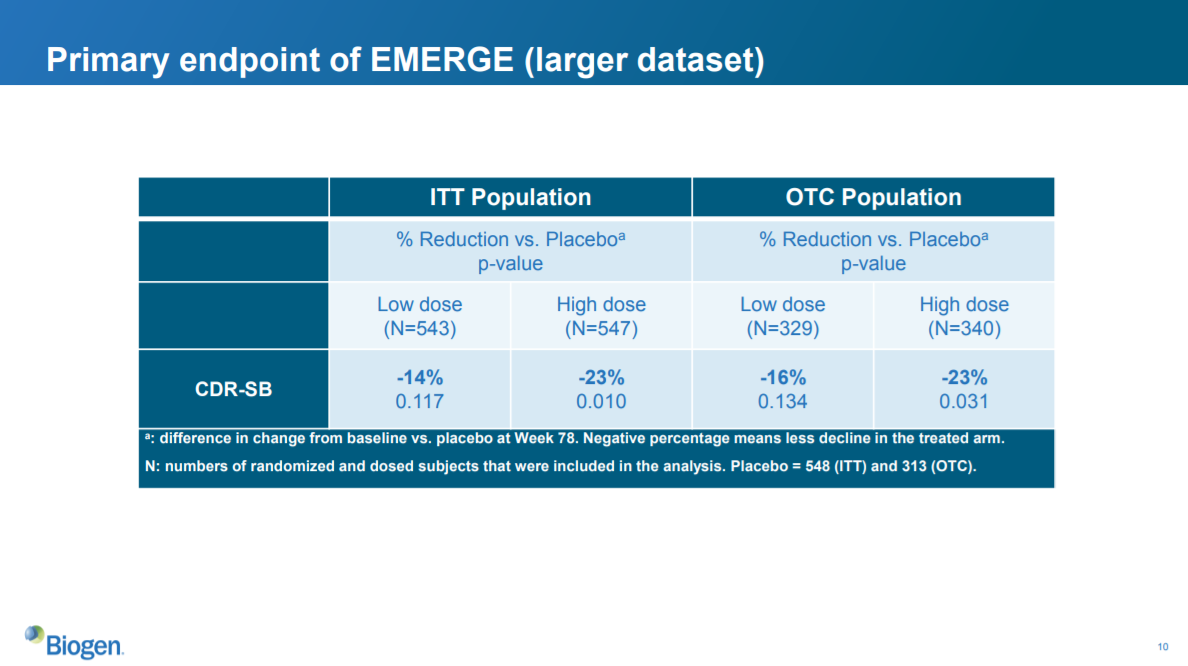
This slide shows how much aducanumab reduced the progression of dementia compared to placebo. The columns (ITT, OTC, low dose, high dose) list the patients included in each subset; the CDR-SB row shows the reduction in dementia scores compared to patients who got placebo.
ITT analysis
ITT means “intent to treat”. This represents all of the patients enrolled in the study. OTC means “opportunity to complete”. This represents a subset of the ITT population that reached a certain study milestone by a certain date. In this case, it represents patients who attended the physician visit scheduled for the 78th week of the study.
The OTC population can be smaller than the ITT population for a number of reasons: patients may drop out of the study due to a safety concern, they may pass away, they may not be far along enough in the study, they may just stop coming in for visits and responding to outreach, etc.
Generally, both ITT populations and a population that receives the treatment as intended are included in an analysis. At first glance, including an ITT population may not make sense -- if patients didn't receive the drug as intended, isn't that just noise? Including ITT analysis preserves the original sample size, reflects a realistic clinical scenario (clinical studies are very controlled environments and may not reflect more general patient behavior), and captures any noncompliance related to drug (if patients worsen on the drug and drop out, you need to account for that). It also protects against bias -- if as a result of patient drop out, one arm has a sicker group of patients than another, then we cannot make an apples-to-apples comparison across arms.
Primary endpoint
The bold percentages in the CDR-SB row represent percent reduction of the group compared to placebo. So the low-dose group in the ITT population decreased their CDR-SB dementia scores 14% compared to the ITT placebo group.
The numbers underneath the percentages represent the p-values. So the aforementioned 14% reduction vs. placebo has a p-value of 0.117, which is not statistically significant at a threshold of 0.05.
The data show that the high-dose group in both the ITT and OTC populations had a statistically significant improvement in dementia scores vs. placebo. This means that the study achieved the primary endpoint. This is good news.
Statistical significance =/= clinical significance
But is this clinically meaningful? A statistically significant result may not always be clinically meaningful. A clinical improvement can be very small but still statistically significant: if a drug showed a statistically significant reduction in the number of times you sneeze per day when you have a cold, but it only reduces sneezes from 14 times a day to 13, is that clinically significant?
I don’t know whether a 20% reduction in CDR-SB is considered clinically significant. I don’t know whether CDR-SB is considered a good scale for measuring dementia. To answer these questions, you’d need to talk to physicians with expertise in Alzheimer’s (often referred to as “key opinion leaders” or “KOLs”). I assume that measuring dementia is an accurate and meaningful way to measure how Alzheimer’s impacts a patient’s quality of life, but in other indications where the relationship between a symptom and quality of life is less clear, it is important to talk to doctors and patients to understand how clinically meaningful a symptom is.
Based on commentary I’ve seen from analysts and investors, it looks like a 20% improvement in CDR-SB is on the low range of what is considered clinically meaningful, so it is a "marginal benefit". However, as there are no effective treatments for slowing cognitive decline in AD, this is still very important for patients.
Beyond the primary endpoint -- supporting evidence
So looking at the statistical significance and clinical significance of the drug’s effect on the primary endpoint is critical in interpreting a study. But we can’t just blindly trust statistical significance of the impact on primary endpoint. Because the p-values were 0.01 in ITT and 0.031 in the OTC population, there is a non-zero probability that the null hypothesis (ie, the hypothesis that placebo and the high dose have the same reduction in dementia scores, ie the drug doesn't work) is true. The positive result could be a false positive. So we look to other data to corroborate the interpretation that the high dose is in fact better than placebo.
One common piece of corroborating evidence is the presence of a dose-responsive improvement on the primary endpoint. That is, does the high dose look better than the low dose? If so, that suggests that the improvement in outcomes is in fact associated with the drug. That seems to be the case here, which is encouraging.
We also look at the secondary endpoints, which are discussed on the next slide.
Slide 11: EMERGE secondary endpoints
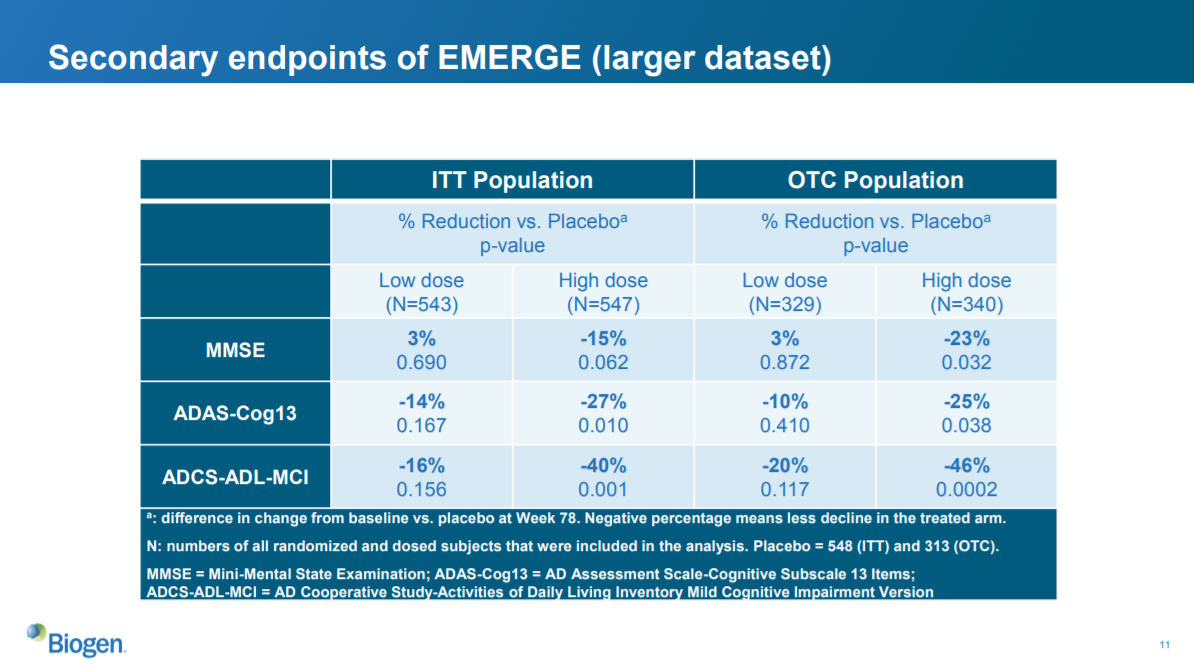
The secondary endpoints are MMSE, ADAS-Cog13 and ADCS-ADL-MCI. These are all either measures of cognitive impairment or ability to function day-to-day (using the phone, going shopping, etc).
We see statistically significant improvement on all of these endpoints except the MMSE in the ITT population, and on all secondaries for the OTC population, for the high-dose group vs. placebo (assuming a significance threshold of 0.05). The low-dose group does not show statistically significant improvements, but shows nominal improvements for ADAS-Cog13 and ADCS-ADL-MCI and nominal worsening on MMSE.
Overall, these data support the primary endpoint in suggesting that the high dose is an effective treatment for Alzheimer’s disease.
Again, I don’t know whether the magnitude of these improvements is clinically meaningful. For an indication that had good standard-of-care treatments, it would be important to understand whether the improvements are clinically meaningful. Otherwise physicians may prescribe an alternative treatment even if your drug is approved. It is also important to consider the safety of the drug. Even if there are no alternatives, if a drug provides only modest benefit but is not safe, it is unlikely to be used.
In this case, the drug appears to have a manageable safety profile (see slide 16), and because there is nothing else for AD, it would likely get significant use.
So the EMERGE study looks good! Based on the data, aducanumab looks like an important treatment for Alzheimer’s patients. Because there are so many Alzheimer’s patients, it looks like a nice product for Biogen.
BUT!
Slide 12: ENGAGE primary and secondary endpoints
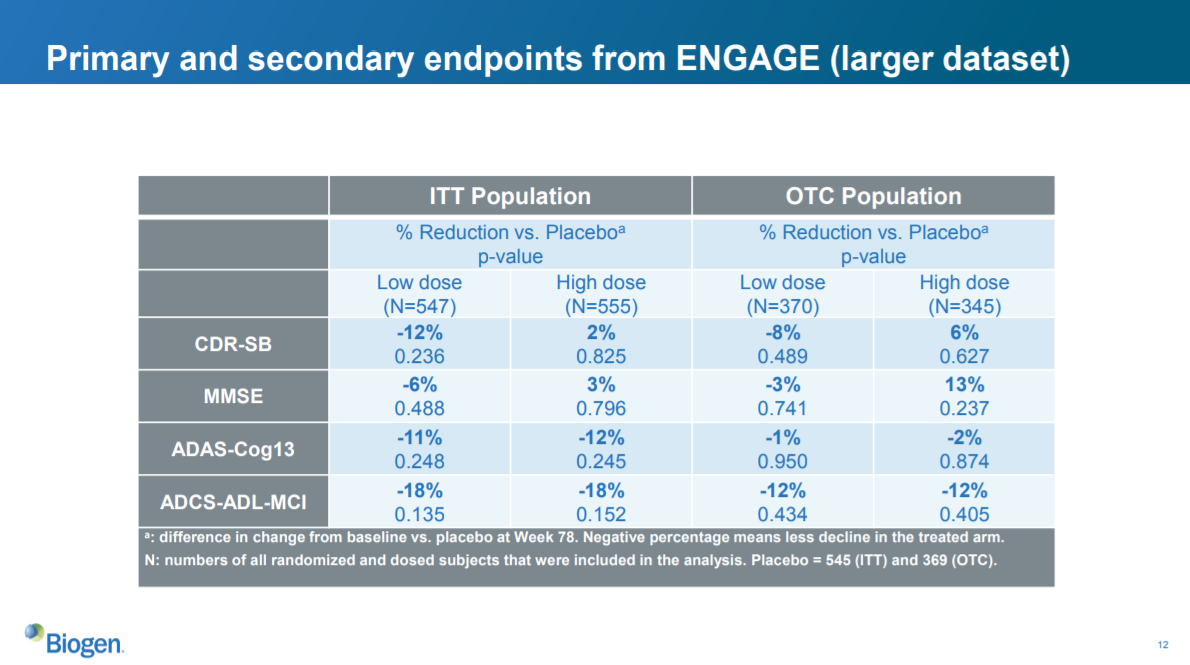
EMERGE looked good. How did the second study, ENGAGE, look?
In short, bad.
Neither the low dose nor the high dose reached statistical significance on any of the primary or secondary endpoints in either the ITT or OTC population. The high dose did worse than the low dose on CDR-SB and MMSE, and both were equally disappointing on ADAS-Cog13 and ADCS-ADL-MCI. The high dose nominally increased dementia and cognitive impairment as measured by CDR-SB and MMSE.
Essentially, the drug did no better than a placebo.
This study essentially refutes the positive EMERGE study. The data here represent the full dataset available to Biogen; when Biogen announced the study failed a futility analysis in March, that failure was based on a smaller dataset that appears to have been even worse. Recall that all other large studies of amyloid beta-targeting therapies in AD had failed, so ENGAGE is consistent with that evidence, while EMERGE is not.
The stock market’s reaction to this data was to cut Biogen’s value by $30B. That implies that the market thought aducanumab was worth over $30B. Probably even over $50B, because the market had priced in some risk that ENGAGE would fail and the drug wouldn’t be approved.
This was seen as the final nail in the coffin of the beta amyloid theory of Alzheimer’s disease. Before EMERGE, essentially every large study of a beta amyloid-reducing therapy had failed to help patients with Alzheimer’s. EMERGE and ENGAGE were supposed to be the best designed studies of this class of drugs yet, and aducanumab was thought to be the best beta amyloid targeting molecule developed. In light of ENGAGE and all the other failed studies, and the “marginal” clinical benefit of the high dose of aducanumab in the EMERGE study, anti-beta amyloid drugs seems unlikely to help Alzheimer's patients.
Let's look at some of the other data Biogen presented, and then we'll jump into their new analysis that led them to change their mind about whether the drug works.
Slide 13: data on amyloid clearance
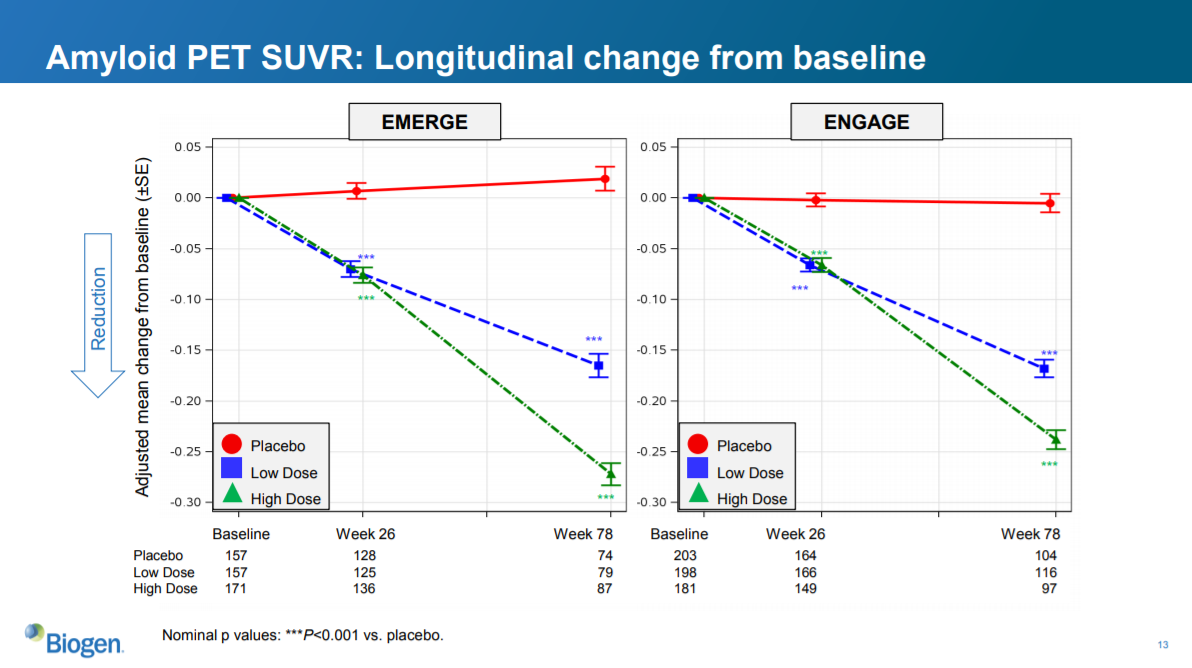
This slide shows the magnitude of reduction in amyloid plaques, as measured by a special type of PET scan, as a result of treatment. Recall that aducanumab is supposed to work by reducing buildup of amyloid plaques in the brain. Because these plaques are thought to cause cognitive decline, reducing these plaques should slow disease progression. At least according to the amyloid beta hypothesis.
As further evidence that reducing amyloid beta doesn’t improve AD outcomes, this slide shows that aducanumab significantly reduces levels of amyloid beta plaques in both EMERGE and ENGAGE. The drug does the thing that it is supposed to from a biological perspective -- reducing amyloid plaques -- but it does not do what it needs to from a clinical perspective -- slowing the progression of AD.
It is worth pausing to comment on this phenemonon: that the a drug's ability to modulate its "target" (in this case, amyloid beta) is a very different thing than a drug's ability to treat a disease. The "target" is a molecule that is thought to be a significant driver of disease. When you “discover” a drug, you are basically optimizing for a molecule with the intended effect on the target, with the constraints that the molecule needs to have drug-like properties (only interact with the target and not other molecules, get to the tissue that it needs to, be safe, be able to be metabolized properly, etc).
Optimizing for a drug that has a specific effect on a target is a very different thing than optimizing for a specific effect on a disease. Scientists can design in vitro assays to measure whether a drug inhibits a target, test millions of chemicals in that system, and then optimize a drug using that system. They can’t throw millions of chemicals into sick humans and see which ones improve disease.
The biggest challenge of drug development is picking targets that are important to disease. The amyloid beta theory is a classic example of this challenge. The scientists who discovered aducanumab appear to have done a phenomenal job creating a drug that reduces buildup of amyloid beta plaques. The clinicians who designed and executed the EMERGE and ENGAGE studies seem to have done an amazing job running a clean study. But the target itself – amyloid beta – did its job very poorly.
A final note on this topic. If you are analyzing a mid-stage study (as opposed to a late-stage study like EMERGE or ENGAGE) that measures a biomarker endpoint (like amyloid beta levels) rather than a clinical endpoint (like reduction in dementia scores), make sure you understand how well the biomarker predicts clinical outcomes. Because a biomarker is often easier / quicker to measure than the important clinical outcomes, in many cases companies will study biomarker endpoints rather than clinical endpoints in mid-stage studies to get an early read on whether the drug is working as intended. But be cautious in assuming that success on a biomarker endpoint will predict success on outcomes endpoints.
Slide 14: data on tau pathology
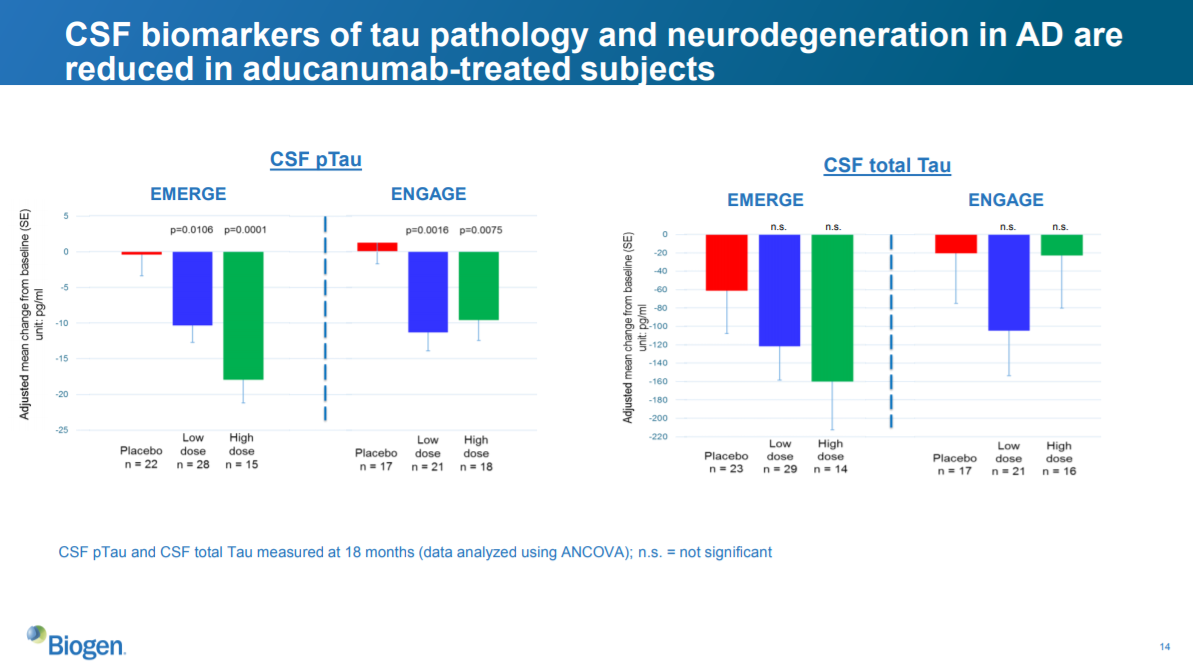
This slide shows ability of aducanumab to improve another biomarker, tau pathology. It looks a bit messier than the amyloid beta data, and, like the primary and secondary endpoints in ENGAGE, shows the a high dose that doesn't seem as good as the low dose. But the sample sizes are quite small here, and I don't know a lot about tau pathology, so I'll move on.
That's it for the clinical data presented. Based on this data, EMERGE showed a seemingly real but perhaps marginal improvement in AD symptoms at the high dose. ENGAGE showed that aducanumab was no better than placebo. The company originally discontinued development of aducanumab based on these data.
But that has changed.
The post-hoc analysis
A post-hoc analysis is a statistcal analysis that was not specified before the data was seen. Post-hoc analyses can be useful: for example, they help researchers generate hypotheses to test in future studies. In earlier AD studies, mild patients responded better to drug than more severe patients. Based on this exploratory analysis, researchers could hypothesize that the drugs might work better in mild patients, and test that hypothesis in a subsequent study.
But drawing conclusions from post-hoc analyses (as opposed to creating hypotheses) is dangerous. Researchers collect many thousands of data points on hundreds of parameters during clinical studies, and there are many ways these data can be analyzed. By mere chance, some of these analyses will look positive. If someone is intent on finding positive data in a huge dataset, they usually can. To prevent any statistical manipulation, FDA requires predefined endpoints and statistical analysis plans to support approval.
To somewhat address the limitations of post hoc analyses, companies can use certain statistical techniques to protect from drawing false conclusions. At the very least, companies can disclose all of their data (or at least all of the relevant data), rather than just showing cherry-picked data, or they can provide justification for why they chose to present a particular analysis rather than others, and show any other analyses they may have done. Doing these things won't enable you to draw conclusions from post hoc analyses, but can at least provide comfort that the researchers are acting in good faith.
If they do not do any of these things, then we can't rule out the possibility that a company is simply data dredging.
Getting back to Biogen: as we saw, the EMERGE data looked pretty good, but ENGAGE was a mess. ENGAGE was so discouraging that Biogen decided to stop development of aducanumab.
But we learned last week that Biogen is resurrecting the program. What happened?
Slide 3: situation overview

This slide summarizes their decision to submit the drug to FDA for approval. Basically, it says that Biogen re-evaluated the data from the study, decided the futility analysis was wrong, and decided to submit aducanumab for FDA approval to treat Alzheimer’s.
What new data led to this decision? Apparently, some patients took the higher dose of aducanumab for a longer period of time, and those patients were not properly accounted for in the futility analysis. When considering a “larger dataset” including these patients, the data show that aducanumab reduced clinical decline in Alzheimer’s patients.
This slide isn’t too clear to me, and seems a bit hand-wavy. But it’s a summary slide, so that’s ok as long as they clarify things and provide more detail in subsequent slides.
One thing I’ll note is that in the first clause of this slide, Biogen notes that it has had discussions with FDA. This does not mean that FDA has agreed that the drug should be approved. It does not mean FDA has agreed to anything. It just means that Biogen has had discussions.
Slide 4: what changed?

In this slide, Biogen provides a bit more context. The salient point for me is that Biogen believes that ENGAGE will be “supportive” of the positive data from EMERGE. This is important because, to approve a drug, FDA typically requires “data from one adequate and well-controlled clinical investigation and confirmatory evidence” (for more on what “adequate and well-controlled” means, see the Code of Federal Regulations here). What Biogen is saying here is that aducanamab checks that box: EMERGE is “one adequate and well-controlled clinical investigation” and ENGAGE is “confirmatory evidence” (remember this is what Biogen is saying and is not necessarily reflective of what FDA thinks).
However, that may not be enough. FDA traditionally requires “at least two adequate and well-controlled studies, each convincing on its own, to establish effectiveness”. It has flexibility to require just one study “if FDA determines that such data and evidence are sufficient to establish effectiveness”.
Biogen’s current data package may check the regulatory boxes, or it may not. It is up to FDA to determine this. Biogen frequently mentions that they have “consulted” and “had discussions” with FDA, which are necessary but not sufficient conditions for understanding whether FDA will require one or two “adequate and well-controlled studies” in this situation. FDA may have said that one study is enough for approval, or they may have just said that Biogen is free to submit an application, and FDA will take a look.
The rest of the slide is a bit vague and, to me, dissatisfying. What is this “additional data” that became available?
Slide 5: "additional data"?

The salient new point here is a bit more detail on the “additional data”. The new data have to do with a "protocol amendment" that enabled more patients in EMERGE (the positive study) to get more exposure to the high dose than in ENGAGE.
What’s a protocol amendment, and why is it relevant? These clinical studies are governed by pre-specified protocols that define who can be in the study, how much drug they get, what “endpoints” doctors measure to see if the drug is safe and effective, and generally how the study is run. These protocols need to be pre-defined to prevent people from fudging with the study design to manipulate results. In some cases, the protocol can be amended for legitimate reasons.
Biogen instituted a protocol amendment that enabled some patients (the ApoE4 patients) to receive a higher dose (10 mg/kg in the amendment vs. 6 mg/kg before the amendment). I haven't looked into the rationale for this amendment, but I assume it's legit.
The next slide discusses the summary protocols and amendment.
Slide 7: the timing of the protocol amendment
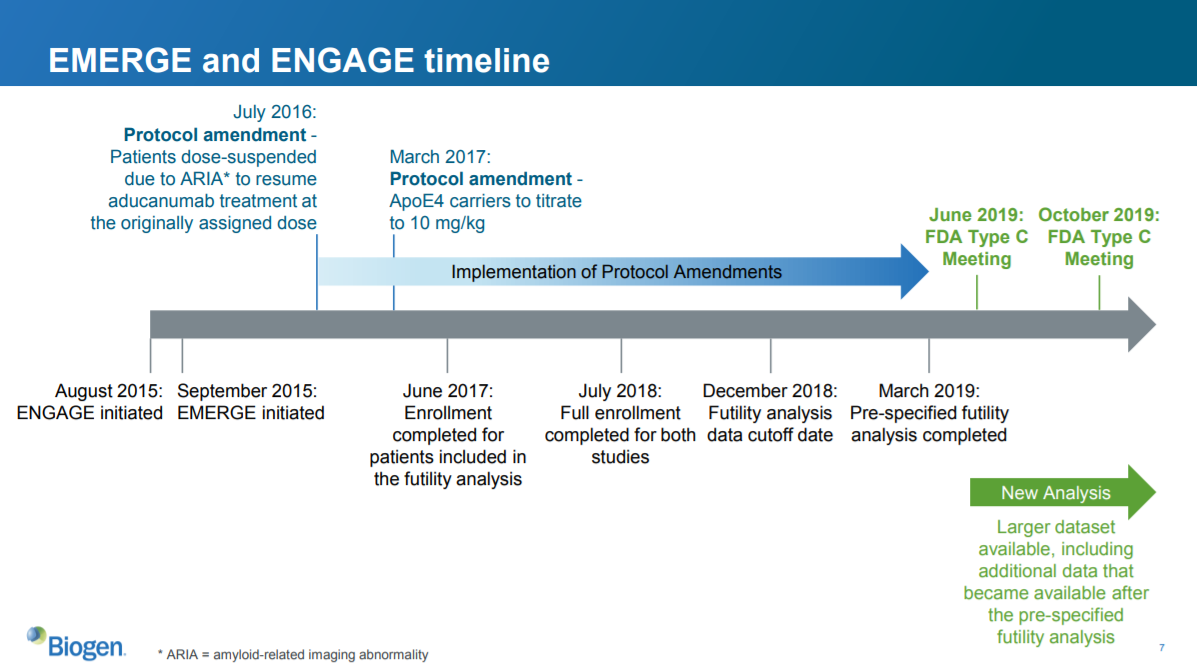
There are two key things here that I can see:
- The timing of the protocol amendment relative to the start of each trial
- The timing of the protocol amendment relative to the completion of the futility analysis and completion of the full study
Both EMERGE and ENGAGE had identical designs, and the protocol amendment applied to both of them. However, EMERGE and ENGAGE started at different times. So the protocol amendment was implemented at different times relative to the start date of each trial.
ENGAGE started one month before EMERGE. The protocol amendment allowing higher doses for ApoE4 carriers was implemented for both studies at the same time. So more ENGAGE patients were treated before the protocol amendment than EMERGE patients. Because the protocol amendment increased the dose for some patients, patients in ENGAGE got the high dose for less time than patients in EMERGE.
The second salient point relates to the timing of the futility analysis. The enrollment for patients included in the futility analysis occurred just three months after the protocol amendment. So most of these patients probably were not on the amended higher dose for very long.
But only a subset of total patients were included in the futility analysis. Enrollment of the complete ENGAGE study did not occur until a year after the enrollment completed for the futility analysis. So the full analysis included more patients who received the high dose for longer, compared to the smaller futility analysis.
I still don't totally follow what happened with a sufficient amount of detail. But on its face, it sounds reasonable that the larger analysis might have included more patients with more exposure to the high dose. And if higher dose leads to better outcomes, maybe the larger dataset may show better outcomes for patients receiving the drug. But there are so many holes and unknowns. And why did the larger dataset -- which we discussed above -- look so bad in ENGAGE, especially the high dose?
Slide 8: the futility analysis revisited

To be honest, I don’t fully follow the statistics discussion on this slide. I'll blame that on the fact that I'm not a statistician rather than any vagueness from Biogen. But my takeaway is that the futility analysis was designed to measure futility by pooling the data from EMERGE and ENGAGE, and then analyzing that data to predict what would happen to the remaining patients in the studies. By pooling the data, you increase your statistical power. However, pooling data only works if both studies were largely identical: otherwise you couldn’t use data from one study to predict the results of another study.
While the studies were designed to be identical, the aforementioned protocol amendment affected them differently because they started at different times. So theoretically this difference could invalidate the futility analysis.
But did it actually invalidate the results of the analysis? I don't know. They don't provide much, or any, data showing how the futility analysis was invalid.
Slide 9: the datasets
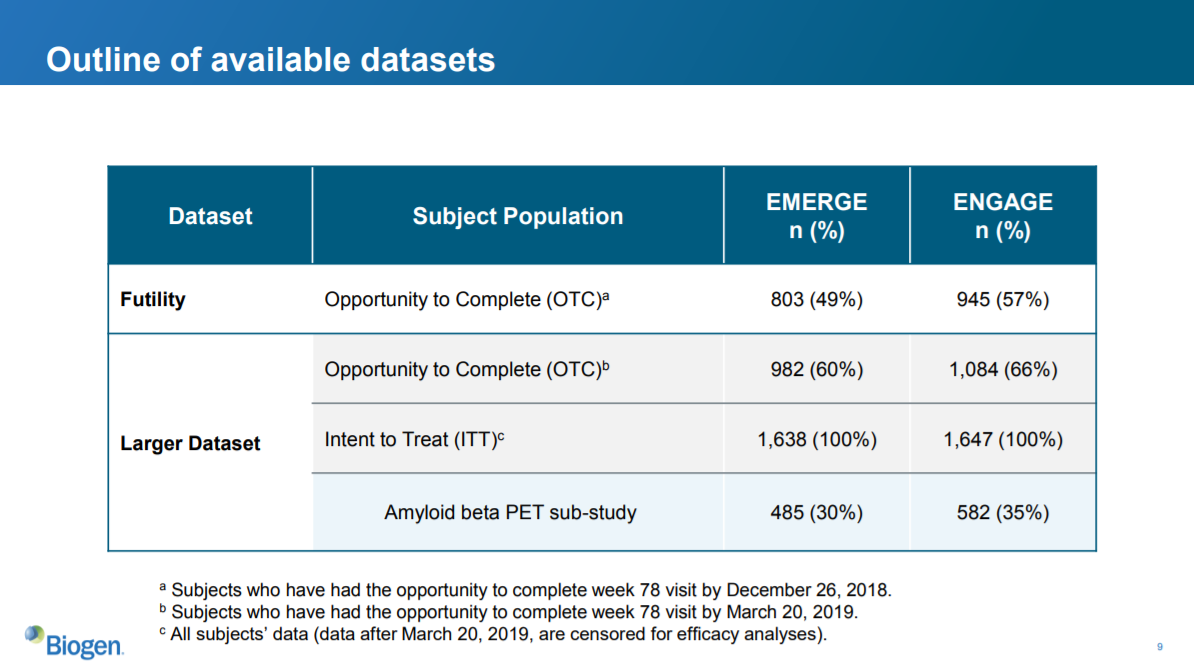
This slide describes the datasets. As we see, more patients in ENGAGE were enrolled at the time of the futility analysis. We also see that 139 more patients were included in the ENGAGE larger dataset than were included in the futility analysis.
So the “larger dataset” for ENGAGE includes 139 patients who presumably had more exposure to the amended higher dose.
To this point in the presentation, Biogen has not actually presented data that supports their new hypothesis. They’ve recapped the existing datasets, discussed the futility analysis and protocol amendment at a high level, and laid out an argument for why the futility analysis is not valid.
The next slide is the first (and only) data presented from their post hoc analysis:
Slide 15: the new data...
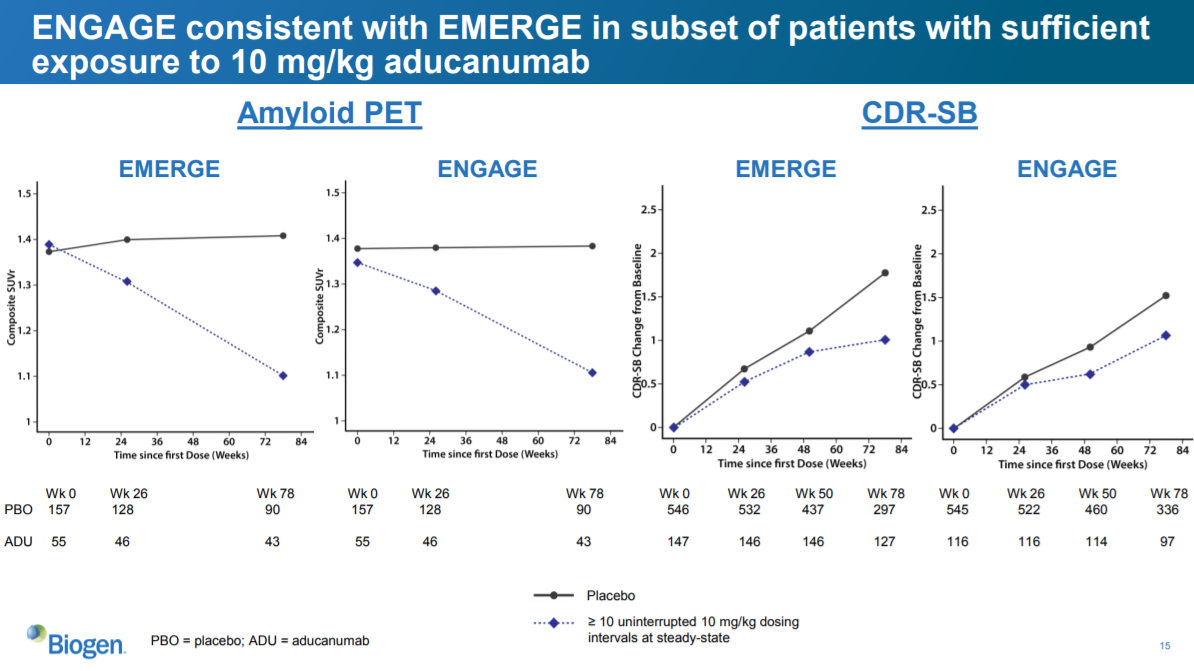
They show two sets of charts here. The two leftmost charts show the reduction in amyloid plaques as measured by PET scan (similar to slide 13). The two rightmost charts show change in CDR-SB from baseline (similar to the tables on slides 10 and 12).
The difference between this analysis and the other analyses is that this analysis only includes patients who had at least 10 uninterrupted 10 mg/kg aducanumab dosing intervals at steady-state.
This reflects Biogen’s prior argument that if you only look at patients who had more exposure to the highest dose, then ENGAGE looks more like EMERGE. And these data…sort of…support that. The amyloid PET data is consistent with slide 13, which isn’t surprising, given the ENGAGE amyloid PET data were similar to the EMERGE data for the larger datasets. The CDR-SB data look more positive in this subset than on slide 12, but that isn’t saying much. Remember, on slide 12 (the full ENGAGE dataset), the high dose did WORSE than placebo. In this chart, the aducanumab dose in ENGAGE looks better than placebo at reducing CDR-SB.
But not by much! The aducanumab dose in ENGAGE here does not look as good as the EMERGE dose. In EMERGE, it looks like there is a 0.7-0.8 improvement in CDR-SB vs. placebo. In the ENGAGE chart, the reduction is only ~0.5. They did not show the actual numbers, so it is hard to tell the exact magnitude of the difference.
There are other limitations as to what we can conclude from these data. For one, the sample size is quite small: there are only 97 patients at the 78-week timepoint in the CDR-SB analysis of the ENGAGE study. Even if this analysis were predefined in the statistical analysis plan (which it wasn’t, as this is a post-hoc analysis), I don’t know that this would be a statistically significant finding.
Another major limitation is that Biogen does not provide any data on the secondary (or any other endpoints) with this new high-exposure-to-high-dose subgroup. That seems like a very logical set of data to include, but they did not show this.
We also don’t know exactly what “>= 10 uninterrupted 10 mg/kg dosing intervals at steady-stage” means. Specifically, we don’t know what “uninterrupted” means. Perhaps the definition of this is clear to someone more familiar with this study or drug. But I don’t know what it means. Does this include people who were “interrupted” because of safety (I think I read that there were some interruptions due to a side effect of the amyloid-beta drugs called ARIA)? Does it include interruptions for people who missed a dose?
This could be innocuous, or it could be relevant. The protocol may have a way of handling people who miss doses. And it may be the case that no patients missed doses. I don't know. But if “interruptions” include people who miss a dose, it may be concerning. Are patients with more significant cognitive impairment more likely to miss doses? At least one of the endpoints (ADCS-ADL-MCI) includes questions about whether the patient missed appointments, so it seems reasonable to ask whether there is a correlation between CDR-SB and missing appointments. If so, only including patients who had “uninterrupted” doses could self-select for patients who were doing better, and thus be a confounding variable. Again, this may be a non-issue, but it’s worth asking.
Another important question is why they used 10 or more uninterrupted dosing intervals as the threshold. Why not 8, or 11? If you use a different threshold, does that make the data look better or worse?
Again, this may be a non-issue. There may be a clear reason for choosing 10 as the threshold.
But this highlights the danger of post hoc analyses, especially when the company does not share the full dataset or their methodology for doing these analyses. If the company only shows us the results of one analysis and does not describe their methodology for choosing this particular analysis, we have no idea if the result is sound, or if they just chose 1 analysis out of 100 that looked good. This is why companies must predefine their endpoints and statistical analysis plans – otherwise it is too easy to massage the data.
It is also frustrating that they’ve presented these data as absolute change from baseline, rather than as a percentage change from baseline, which is what they showed on slides 10 and 12. It would be nice to see whether the change in CDR-SB score here was greater than the change in CDR-SB that was seen in EMERGE. If the company’s hypothesis that more exposure to the higher dose results in more CDR-SB reduction, we’d hope to see that the change in CDR-SB shown on slide 15 is greater than that shown on slides 10 and 12.
But we can’t make that comparison. To calculate the percent change from baseline, we need to know the value at baseline, and then the value at the final timepoint. The baseline CDR-SB isn’t shown on slide 15 – just the absolute value of the change from baseline. So we can’t compare the magnitude of the change in CDR-SB in the "new" high-exposure-to-high-dose group to the 23% reduction in CDR-SB in the EMERGE high dose group vs. placebo.
Presenting the data on slide 15 in a way that can't be compared with earlier CDR-SB data isn't necessarily a bad thing, but it is frustrating and prevents us from conducting a potentially useful comparison.
There may be good reasons for not including these analyses. But I don’t think we can rule out the possibility that the data just isn’t that good.
Incentives
Aducanumab, if approved, could be the biggest selling drug ever. Some analysts estimate $10B+ in annual sales. That would put it up there with Humira, the best selling drug of all time ($19.9B in 2018 sales), Harvoni ($14B in 2015 sales, although Harvoni sales have dropped dramatically since then after the arrival of cheaper competitors), and Lipitor ($14B in peak sales in 2006). When the futility analysis of the ENGAGE study was announced in early 2019, Biogen lost $30B+ in market cap overnight. Because that $30B included a discount – the drug was not yet approved, so there was some probability of failure – the drug is worth more than $30B, potentially much more than $30B.
The drug is critical to Biogen. Biogen’s current market cap is about $55B (as of the end of October 2019). Biogen’s market cap was approximately $40B before the company announced it was going to submit aducanumab for approval after all. So aducanumab is potentially worth more than the rest of the company combined.
Beyond just money, I’m sure that aducanumab is very important to many Biogen employees, many of whom have likely devoted much of their careers to developing this drug. And as I mentioned earlier, it seems like a great molecule and a very worthwhile project – the amyloid beta theory was the best theory scientists had around treating Alzheimer’s and was supported by a sizable body of evidence, and from what it looks like Biogen has developed a very nice molecule for reducing amyloid levels. Drug development is risky, and the fact that a drug ultimately fails does not take anything away from the work scientists did to develop the drug.
And if the drug does in fact work, it would be very valuable for patients. There are currently no disease-modifying treatments for Alzheimer’s, and even if Biogen’s drug provides only modest improvements, those could be unmeasurably important for patients and their families. More importantly, it would stoke the enthusiasm of drug companies to develop better drugs.
So I can understand Biogen wanting to submit this drug for approval even if it seems unlikely to get approved. Because the expensive studies are all done, submitting for approval would be a tiny cost compared to the potential value of the drug -- it may cost less than $100M to submit the application. Even if there is a less than 1% chance of approval, it could make financial sense to submit. And it they've come this far, it seems reasonable for them to have an honest conversation with FDA about whether this drug might be helpful for patients.
But if Biogen is being misleading with their data, and trying to push a drug that doesn't work through FDA so they can make a bunch of money, that is clearly unethical and irresponsible. The industry (including investors) should keep a close eye on this and push for an open and transparent discussion of the data.
Benchmark investor performance
See which funds will survive market turmoil with deal-level returns, portfolio overviews, and performance metrics for thousands of public and private biotech investors covering hundreds of billions of dollars of investment.
You may also like...
The world's most expensive drug? A case study of Zolgensma
A synthetic biology paper, in layman's terms
Valuing drugs and biotech companies
Did you enjoy this article?
Then consider joining our mailing list. I periodically publish data-driven articles on the biotech startup and VC world.