Basics of drug development for those unfamiliar with the industry
Richard Murphey, 4/13/2018
The drug development process is one of the most complex, technical, expensive and highly regulated industrial processes in the world economy. It is also very important for society, as it is the source of nearly all new medicines our world receives. The cost of this process is a, if not the, major reason for the high price of drugs. For anyone trying to solve the problem of high drug prices without stifling innovation, understanding how drugs are developed is a key piece of foundational knowledge.
This post will attempt to provide a high level orientation to the process and introduce some technical terms while hopefully being accurate and understandable for those unfamiliar with the field.
If you have corrections / suggestions for improvement, please let me know!
Breaking down the drug development process
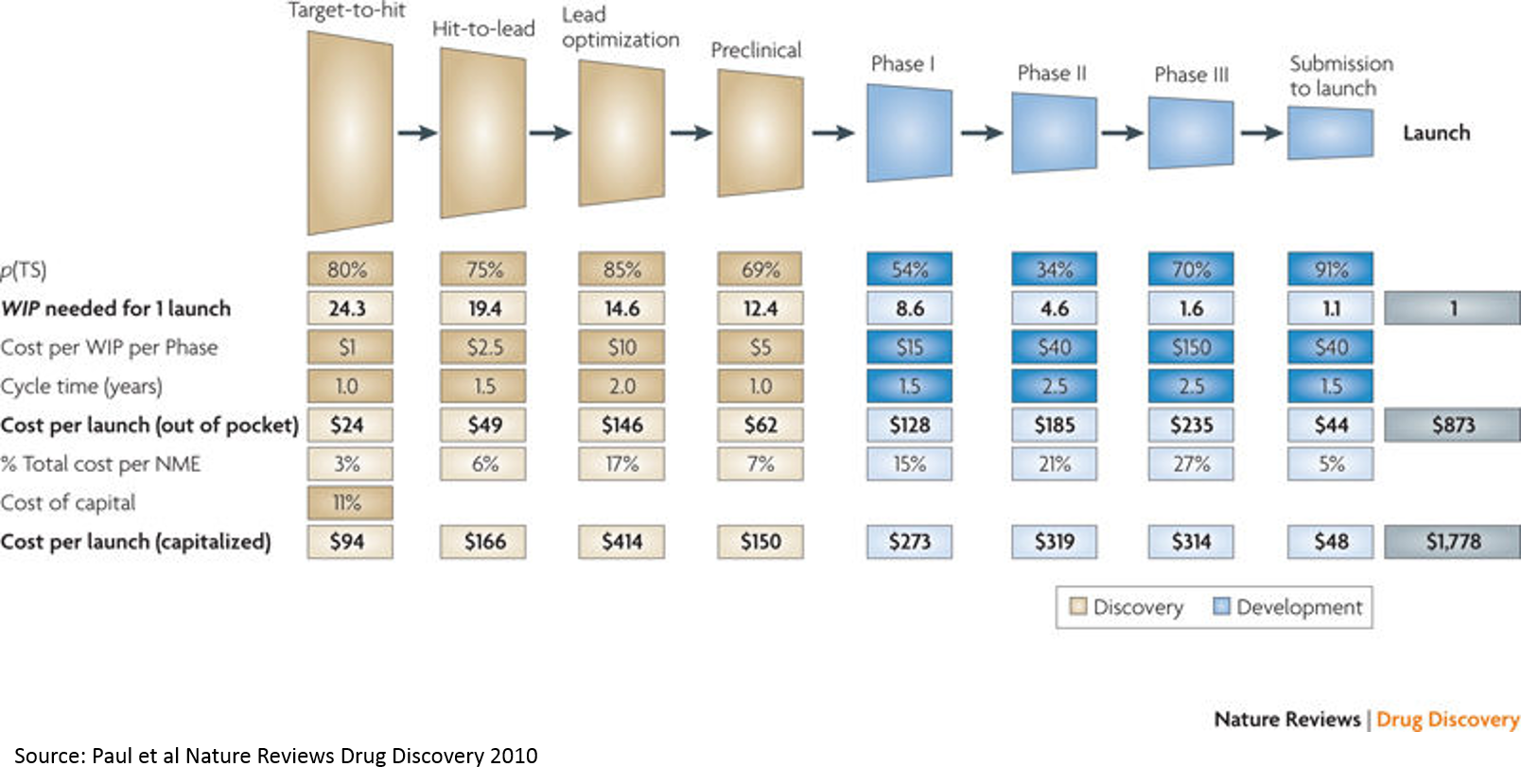
The above paper is a commonly cited source for the cost of drug R&D (it has been updated several times since 2010 and there is considerable and heated debate about methodology, but the above chart from the 2010 paper is a useful graphic). While there are many competing approaches for calculating this cost, this serves as a good starting point.
For context, drugs are generally either 1) small molecules or 2) large molecules. Drugs work by blocking, augmenting, or otherwise modifying the activity of a molecule (usually a protein) involved in a disease process -- this molecule of interest is referred to as a "target". Generally drugs have one intended target, although many if not most interact with additional molecules.
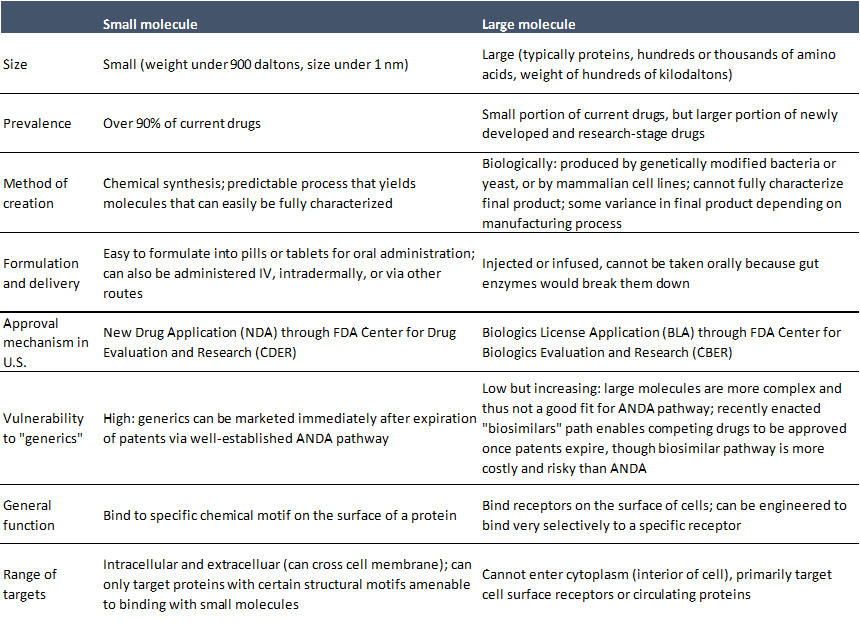
The following describes the process of developing small molecules. The process for developing large molecules is philosophically similar with some major and minor differences, but these are beyond the scope of this discussion. I think some knowledge of these steps is essential to understanding drug development, although I'd encourage anyone who is interested to read more (for a good overview with a bit more detail, see here) or talk to scientists!
-
Target discovery and validation: This does not appear on the above chart, but is the process of identifying a putatively druggable target that plays an important role in a disease, and then performing experiments to verify the target's hypotheized role in the disease of interest. The only way to definitively know if a target is involved in disease is to test this in humans, but we cannot test unproven compounds in humans, so target validation is done in animals and in vitro (this is also much cheaper than doing human studies). There are many ways to validate a target, including binding the target with other molecules and observing the effects, genetically blocking or augmenting the target and observing the effects, or analyzing the molecular pathways downstream of the target.
-
Target-to-hit: This is usually accomplished through a process called high-throughput screening. Researchers develop an assay measuring ability of molecules to bind to a target, or to effect some desired functional outcome in a cell based assay or disease model. They then screen huge libraries of compounds (often in the millions of compounds) to see which ones "hit" the target. Big pharma companies have large chemical libraries and fully automated high throughput screening labs. This can also be outsourced to CROs (contract research organizations), which is a good option for small companies.
One advantage of high throughput screening is that you don't need to know what kind of drug might hit the target: you just screen a ton of compounds and see what sticks. If you have some prior knowledge of the kinds of chemicals that might interact with the target, you can do a more focused screen. Computational ("in silico") tools can help with this.
The primary assay may be one that measures the binding affinity of a compound to the target. Additional assays are developed to ensure the compound has the desired biologic activity, confirm the observed ability to bind to the target, or otherwise narrow down the list of potentially promising compounds. Assay development is a core part of this process. A good assay needs to be reproducible, reliable, low-cost, quantitative, and representative of the biology of interest subject to the prior constraints.
-
Hit-to-lead: After identifying hits in the primary and secondary assays, researchers need to triage the hits to create a shortlist of the best candidates for further testing. The process of developing a hit into an optimized "lead" compound (a compound that will be advanced further into development) is quite expensive and time consuming, so it is important to be selective about what compounds move forward.
This filtering is done with a combination of computational modeling techniques, experimental assessments of key parameters determining whether the compound has "drug like" characteristics (gets into the circulation and stays there long enough to get to the target, is selective for the target, doesn't interfere with metabolism / healthy function, is not toxic, etc.), and scientific judgment. Researchers try to include compounds with a variety of chemical structures to ensure they have a broad set of potential leads.
Researchers then evaluate each "hit series" to try to produce more potent and selective componds to advance into animal models. This consists of an intensive examination of the structure-activity relationship (SAR -- what parts of the compound are responsible for which specific biochemical activities of the compound) analyzing the structure of the target as well as the hits. This is combined with a series of assays measuring selectivity (does the compound hit just the target, or other proteins), functional assays (what happens in a model of disease when it hits the target), and primary assays in different species. Other assays aim to understand how the compound might act in the body (solubility, permeability, how it is absorbed into the bloodstream and distributed, how it is metabolized, how it is excreted). Compounds are further filtered based on this information.
The remaining candidates are often evaluated in animal DMPK models (drug metabolism pharmacokinetics). Animal studies are more expensive than in vitro screens (screening a million compounds in mice would require a million mice), so this is generally a later-stage filter. These studies aim to discover where the drugs go in the body, how much of them circulate in the blood, and how long the drugs stick around.
-
Lead optimization: While the goal of prior stages is to filter a long list of compounds into progressively shorter lists until finally selecting a lead compound, the goal of the lead optimization stage is to engineer the lead compound to have the best possible therapeutic effect with lowest possible safety risk. This is a costly and time-consuming phase, generally more costly than all the prior work combined.
If any flaws or shortfalls of the compound were identified previously, chemists aim to address these by modifying the chemical structure of the compound. Researchers iteratively modify and test molecules using the same assays as above in addition to other assays.
New assessments at this stage often including genotoxicity testing (does the compound damage DNA), pharmacokinetic / pharmacodynamic (PK/PD) studies (where do drugs go in the body and what effect do they have in the body), dose linearity studies (do increased doses have linear increases in PK / PD parameters) and multiple dose PK studies.
-
Preclinical: After the lead optimization phase, a compound enters the final stages before it can be tested in humans. A major goal of this stage is to assess the safety of a product and determine which doses to use in initial human studies. Some of the studies mentioned above can fall into this bucket as well (genotox, further PK/PD testing).
A core part of this process is toxicology studies. Toxicology studies basically entail giving large amounts ("the tox is in the dose") of a compound to animals and observing what happens. Researchers try to find a maximum tolerable dose (MTD), a no-observed-adverse-effect-level (NOAEL) and other metrics that inform the selection of initial human doses. Generally companies must perform tox studies in two species (usually a rodent and a non-human primate or other large animal), and perform exploratory tox studies as well as more rigorous multiple-dose GLP-compliant (GLP is "Good Laboratory Practice", a set of regulations related to quality controls for preclinical studies) tox studies. The duration of tox studies is determined by the intended duration of dosing in humans, adjusted for the difference in lifespan between humans and animals used in tox studies. Depending on the compound and indication, further studies may be required.
In addition to tox studies that look at commonly occurring harmful effects of very high doses, companies also perform "safety pharmacology" studies to predict rare harmful effects that occur at normal doses.
In order to initiate a human study in the U.S., companies must file an Investigational New Drug (IND) application with the FDA. If FDA does not have any concerns within 30 days, the company can initiate human studies. Companies can request Pre-IND meetings with FDA through a formal mechanism to understand what FDA may require. To improve the quality of IND submissions, FDA offers detailed guidance regarding content on its website, including for nonclinical safety studies. These are a great resource for those who wish to learn more about the regulatory process and drug development.
Another important parallel workstream relates to Chemistry, Manufacturing and Controls (CMC). Scaling up manufacturing of a drug to supply in vitro studies, then early animal studies, then tox studies and progressively larger human studies is a complex undertaking. Formulation development (formulating the active pharmaceutical ingredient (API) into a pill, patch, tablet, injection, etc) is also complex and expensive. While manufacturing processes for early in vitro and animal studies is not too regulated (compared to other aspects of the drug dev process), companies must comply with Good Manufacturing Practice ("GMP") for human studies.
Increasing quality and regulatory / compliance standards is one reason why scaling up manufacturing is challenging, but another is the sheer scale required. To go from initial batches for in vitro work to scale for tox studies can be a jump on the scale of multiple orders of magnitude, and getting to scale to support human studies can be another multiple-orders-of-magnitude jump. Scaling up to tox capacity can cost a few million in some cases.
-
Phase 1: This is the first stage of human testing. These studies are typically small and short. The goals are to determine the safety of the product and the doses to be used in subsequent studies, often by starting with a single low dose in a few patients, and then gradually increasing the dose to a target level provided there are no safety signals, and then dosing patients with multiple doses. For some indications, you can study measures of a drug's effectiveness to treat the disease of interest, often using objective surrogate "biomarker" endpoints, although any findings at this stage are very preliminary and are tertiary goals of the study.
-
Phase 2: This stage is generally the riskiest / most rewarding of all stages of drug development: human proof of concept. While Phase 1 is a preliminary assessment of safety, Phase 2 is a preliminary test of effectiveness. These are larger, longer studies, often with a placebo or another drug as a control, and are generally randomized and blinded.
Sometimes companies will study "surrogate" endpoints in Phase 2 rather than "outcomes based" endpoints. Surrogate endpoints are predictive of outcomes, but can be measured in a shorter timeframe. For example, a company developing a cancer medicine might study reduction in tumor size in Phase 2. The ultimate goal of the treatment is to prolong life (measured by "outcomes" endpoints like improvement in "overall survival" versus a control arm), but to measure this requries larger trials that last years. Measuring a predictive "biomarker" endpoint in Phase 2 allows companies to reduce risk with comparatively less investment. Biomarkers are also used to stratify patients to ensure a more homogenous patient population is studied, which can enable smaller studies and increase likelihood of success. However, using biomarkers as measures of safety or toxicity (rather than just to determine which patients are enrolled in a study) does not increase probability of success, although it may shorten length of studies.
The success rate in Phase 2 (about 30% per a commonly cited study) is the lowest of all steps in the process. It can cost over $100M to get a drug from discovery through Phase 2: this high cost + low success rate is the single biggest reason why drug development is so expensive, and consequently why prices are so high.
-
Phase 3: These "pivotal studies" are the true test of a drug, and largely determine whether FDA will approve the drug. These studies are larger, measure outcomes endpoints in addition to biomarkers and other endpoints, and evaluate safety of the drug when used in large populations over long periods of time.
These studies are the most expensive in dollar terms, although success rates are higher than Phase 1 or 2 because risk has been reduced through prior clinical work.
FDA generally requires two large, well controlled Phase 3 trials, although this can vary depending on the particular product and indication.
-
FDA Approval: After completing its Phase 3 studies, companies will submit a New Drug Application (NDA, for small molecules) or a Biologics License Application (BLA, for large molecules). This is a massive data submission. FDA generally reviews this over 6-9 months. FDA will set a PDUFA date, a target date for their approval decision, although these often change. Sometimes FDA will convene an Advisory Committee of experts to provide feedback on certain drug submissions. The minutes of these meetings are published and are very informative, and often move markets.
Analyzing the costs of drug development
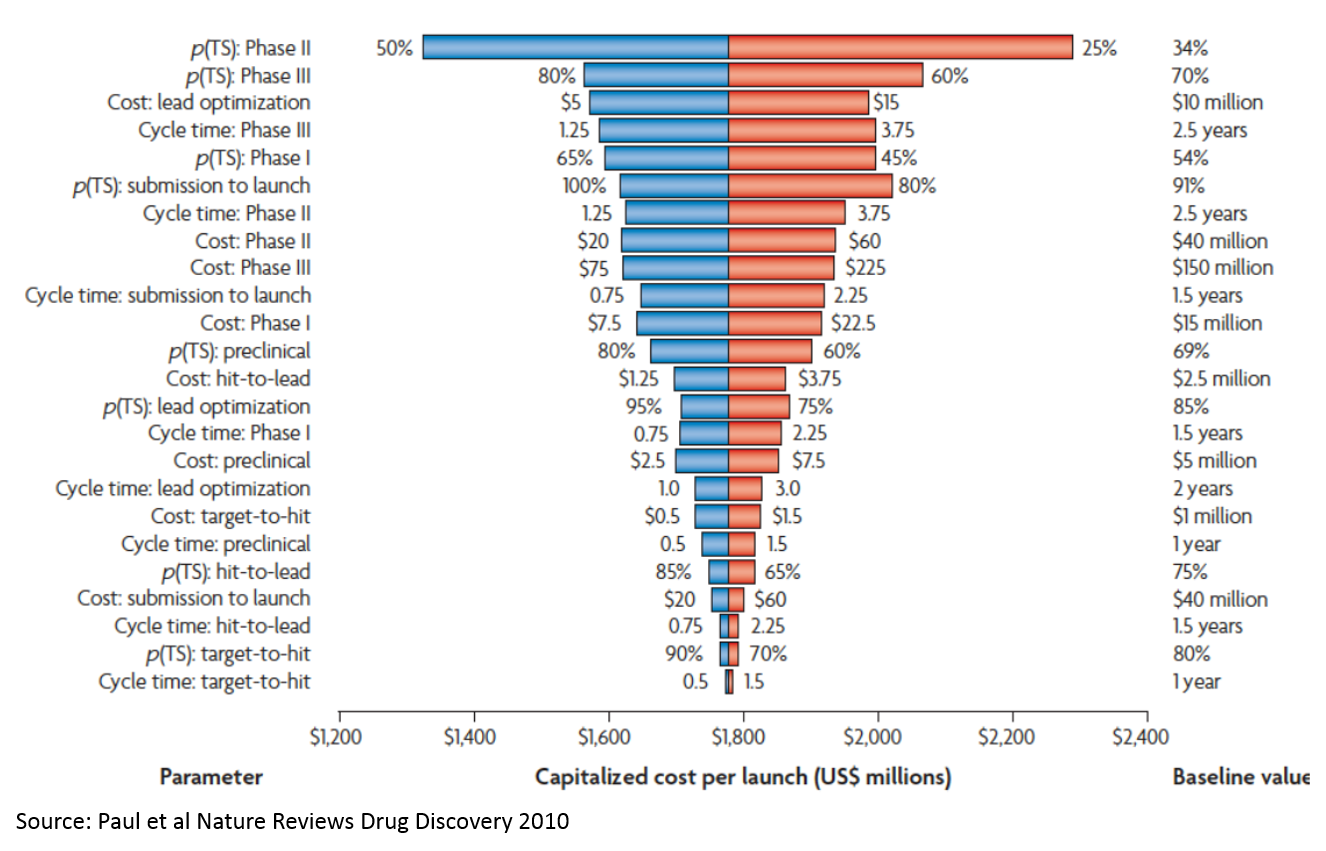
If you add up the costs in the "Cost per WIP per Phase" line, you only get about $265M. But the total cost on the bottom-right is $1.8B (remember, these are 2010 figures, and today's figures are higher due to inflation but mostly Eroom's Law; the most recent estimate from Tufts is $2.6B / approved drug). So where does this $1.5B in "magic" cost come from?
That's the cost of failure. The most significant drivers of this cost are:
- P(TS) Phase 2: the probability that a drug succeeds in a Phase 2 study. Phase 2 studies are usually the first studies to robustly test whether a drug works in humans. Preclinical studies test whether drugs work in animal models, or in the lab, but these studies are very poor predictors of whether drugs will work in humans. Phase 1 studies generally just test which doses are safe to use in humans to inform the design of later stage studies. So you have to invest a lot of money just to get to the point where you are really testing whether your product works.
- P(TS) Phase 3: Phase 3 studies are basically larger confirmatory studies that a drug works and is safe. As you can see, the probability of success is much higher than Phase 2 (which explains why the probability of success in Phase 2 is a bigger cost driver), but Phase 3 studies are more expensive.
- Cost: lead optimization: This is the process where you take a molecule that seems like it works in the simplest lab models, but doesn’t really have what it takes to be a drug. It may be toxic, or its activity may be very nonspecific (so it works against the molecule of interest to a disease, but also messes with a lot of other molecules that could impact healthy function), or it just may be really hard to manufacture at a consistent level of quality. This chemistry work is incredibly time consuming and costly, and requires a specialized skillset that is mostly science but part art. I'm not a chemist, so if you want to learn more, talk to one!
Note that high throughput screening (target to hit) is at the very bottom. “Expanding the funnel” or filtering better at the very top doesn’t really move the needle. This is important, as many of the initial applications of AI to drug discovery relate to “virtual screens” that operate at this stage of the process. While using AI at this stage is appealing because there is a lot of data and the analysis is more straightforward, adding the most value from a drug development perspective would require solving tougher, but more valuable, drug development problems. While predicting human effectiveness with better analytics is not possible yet with just deep learning or other modeling tools, there are clever ways to better predict effectiveness of drugs in human studies by using informatics to complement targeted biological hypotheses. If you can make a dent in this problem, it will be so valuable that it will more than make up for the high cost and technical complexity. Strategies that lower the cost of lead optimization may be a better middle ground: not as technically complex, but still very valuable.
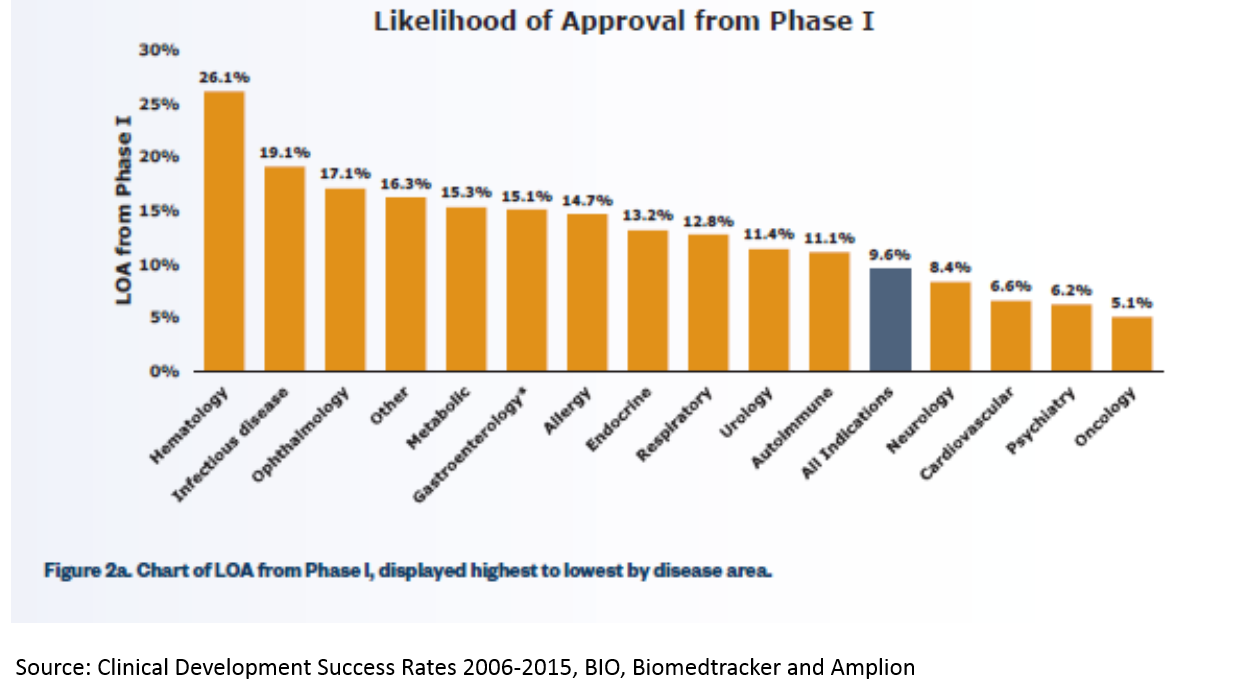
Success rates by disease area
Since clinical success rates are such an important driver of the cost of R&D, it's worth it to take a closer look.
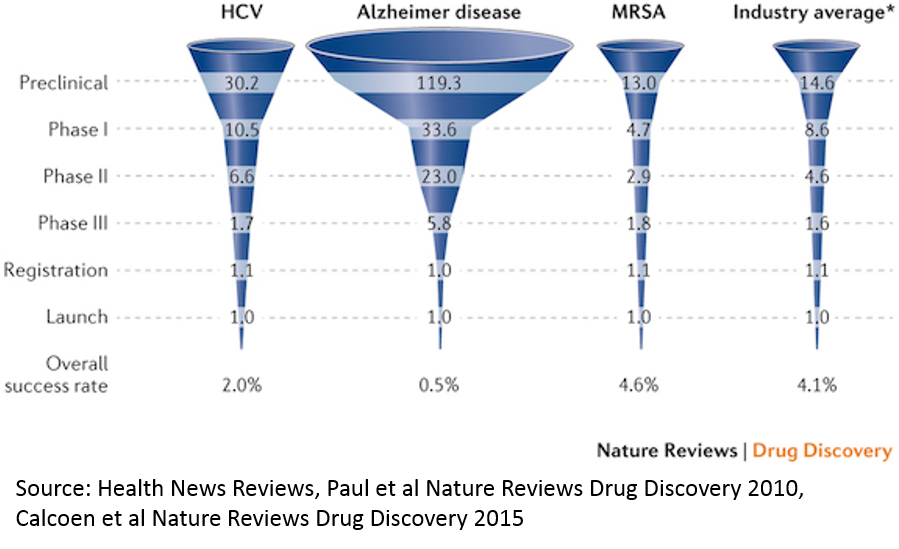
This particular chart highlights some of the highest and lowest success rates in R&D. Generally, anti-infective drugs have the highest rates of clinical trial success, and neurological / psychiatric disease have the lowest. In general, this is because there are better early-stage models for predicting effectivness: if we know a disease is caused by a particular bacteria (like MRSA) or virus (like in Hepatitis C), we can be pretty confident that if we kill the pathogen in question, we can fix the underlying cause of the disease. It's easy to test whether a drug kills a pathogen in a lab, and those cell culture experiments are generally relatively good predictors of whether the drug will work in humans (though still not great per the chart, and also are not great predictors of drug safety).
Compare that to something like Alzheimer's. We don't even understand the biology of the disease well. We also don't have any good preclincial models for the disease: animal brains are very different than human brains, and we can't grow human brains in a dish (though we're making progress with stem cells). So even if we understood the biology of the disease, it would be hard to study whether drugs have a good chance of working in preclinical studies. Another issue is that most drugs can't get into the brain easily, so that restricts the universe of compounds that could be used as drugs, but that doesn't seem to be the rate limiting step, and companies are making progress to address this.
For a more comprehensive look at clinical development success rates by disease and stage, see here. A preview of the data in that report:
Reducing the cost of drug development is not just an existential issue for the biopharma industry, but an important issue for U.S. society. High drug prices are certainly a major issue (although prescription drugs only account for 10% of U.S. healthcare costs; hospitals account for over 30% and physician services for 20%), but I'd argue that the more significant consequence of the high cost of drug development is that we simply don't get new treatments for the most serious public health challenges. Of the ten leading causes of death in the U.S., only cancer receives a significant share of drug R&D investment and new treatments. Heart disease, stroke, respiratory disease, Alzheimer's, mental illness and others are all but abandoned, largely due to the high cost of drug R&D. Addressing this issue is not easy, but innovation is key.
I conclude by linking to a really great summary of a really great paper for contextualizing where we should invest to improve drug development. The gist of the paper is that we've seen an exponential increase in the cost of drug development despite massive improvements in efficiency at key steps of the drug development process. Their conclusion is basically that the disease models we are using are not great predictors of whether drugs work, so massive increases in efficiency just yield larger quantities of not great data. The most predictive disease models have long since been exploited and used to develop good treatments, and now all that are left are the disease models that don't work well. A really provocative conclusion is that a 0.1 absolute change in the correlation coefficient between a disease model and clinical outcomes in man can offset 10-100x improvements in efficiency of executing the disease models. So investing in automated, high throughput labs, or AI-driven virtual screening of millions of compounds, generates a really poor return if the assay you're screening against is bad. Biopharma is really all about biology.