A synthetic biology paper, in layman's terms
by Richard Murphey
In this post, I’ll summarize a synthetic biology paper in layman’s terms. The goal is to spread awareness of some amazing bioengineering research. It is an exciting time to be in biotech, but the field gets comparatively little attention.
I’ll also touch on how I’d think about the commercial potential of the tech, though this is not meant to be an investment analysis.
I'm not a scientist, though I've worked in biotech my whole career. Reading papers, and asking tons of questions to my scientist friends, has been one of the most effective ways for me to learn about science. But getting through papers as a non-scientist is daunting. This post tries to make the paper more approachable and less jargon-heavy without losing too much detail 1.
The main audience for this is technically minded people who are interested in bioengineering, but are not biologists. If you’d just like a high-level overview of the tech but are less interested in the details, you can just read until the “Creating, testing and optimizing RASER” section.
The paper
The paper, from Michael Lin’s lab at Stanford, was published in Science this May. A friend of mine who is in Michael’s lab, Vandon Duong, helped walk me through the paper.
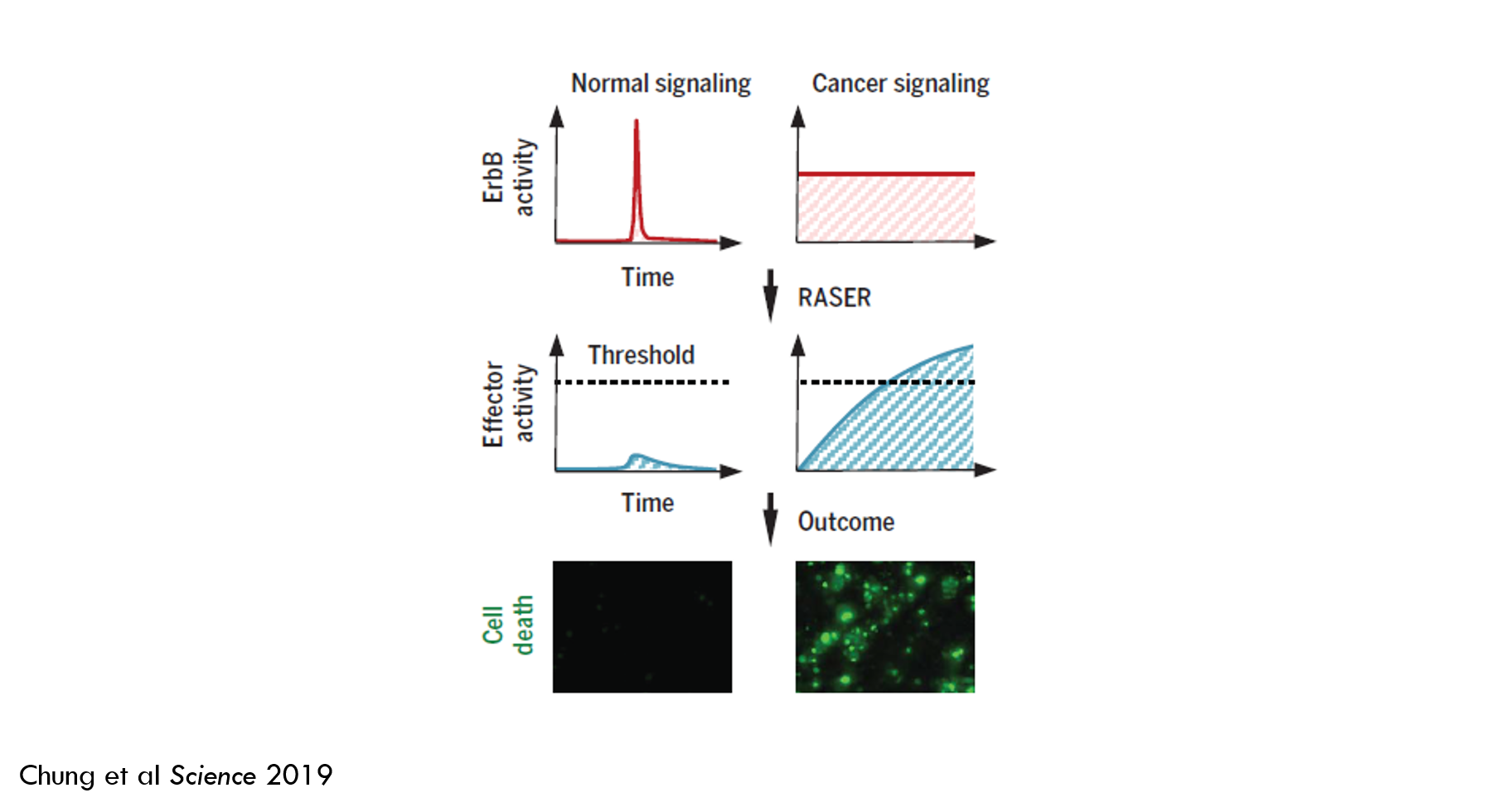
In brief: they developed a tool called RASER (Rewiring of Aberrant Signaling to Effector Release) that hijacks a signaling pathway that causes cancer and rewires the pathway to instead kill cancer cells. A “signaling pathway” is a way for cells to respond to their environment. Generally, a molecule on the surface of a cell (a “receptor 2”) detects a molecule outside of the cell (a “ligand”), and then sends a signal that changes the cell’s behavior. RASER detects when this signal is sent and then releases an “effector” molecule. When the amount of released effector crosses a certain threshold, the cancer cell dies.
The paper describes how the authors engineered and tested the system. It’s a nice example of the current state of the art in synthetic biology. It also showcases some common molecular biology techniques and introduces basic concepts in cancer biology.
Brief description of RASER
This paper describes using RASER to target the ErbB signaling pathay. ErbB is a family of proteins that send signals that cause cells to proliferate. Overactive ErbB signaling can lead to rapid, uncontrollable cell growth. In other words, cancer 3.
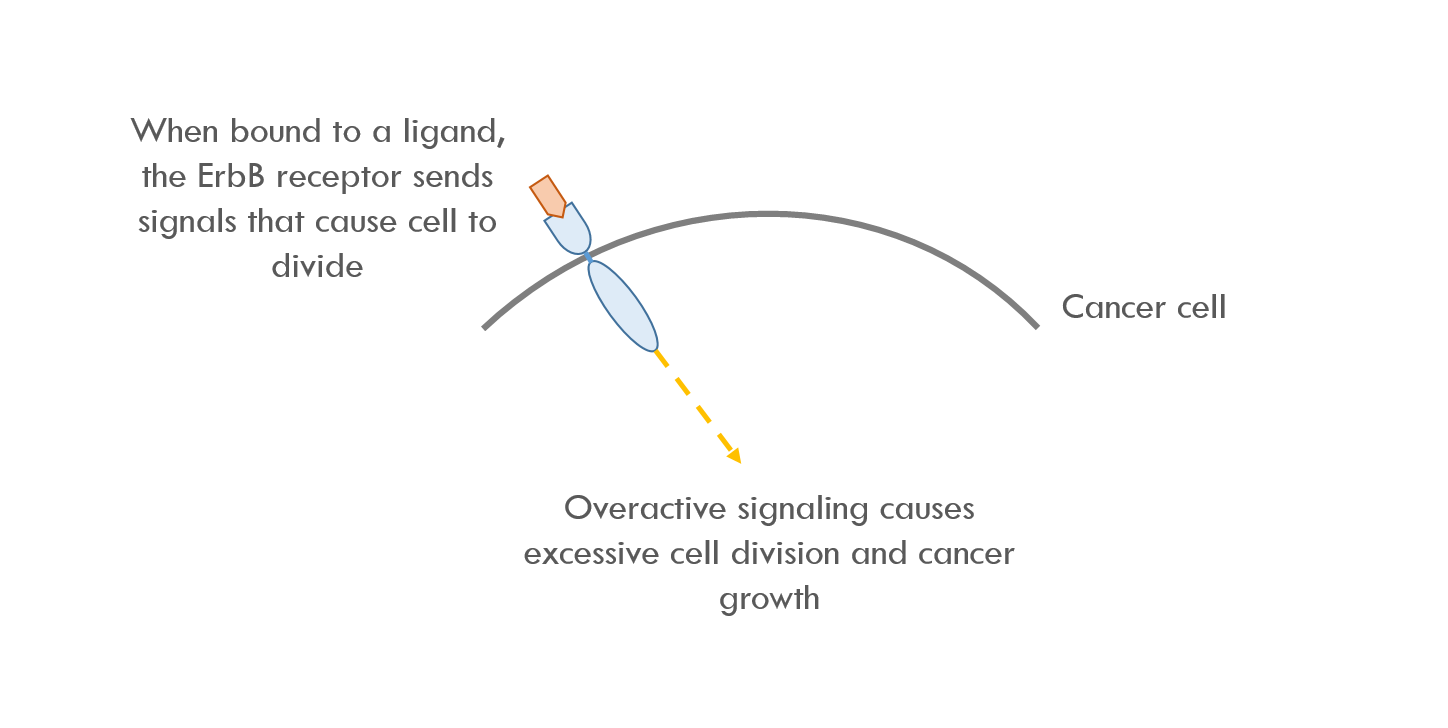
RASER binds to the intracellular part of ErbB, which is the first node in the ErbB signaling pathway. RASER detects when ErbB is activated and then releases a programmable payload. For example, RASER can release a payload that causes cell death, or increases / decreases expression of a specific protein.
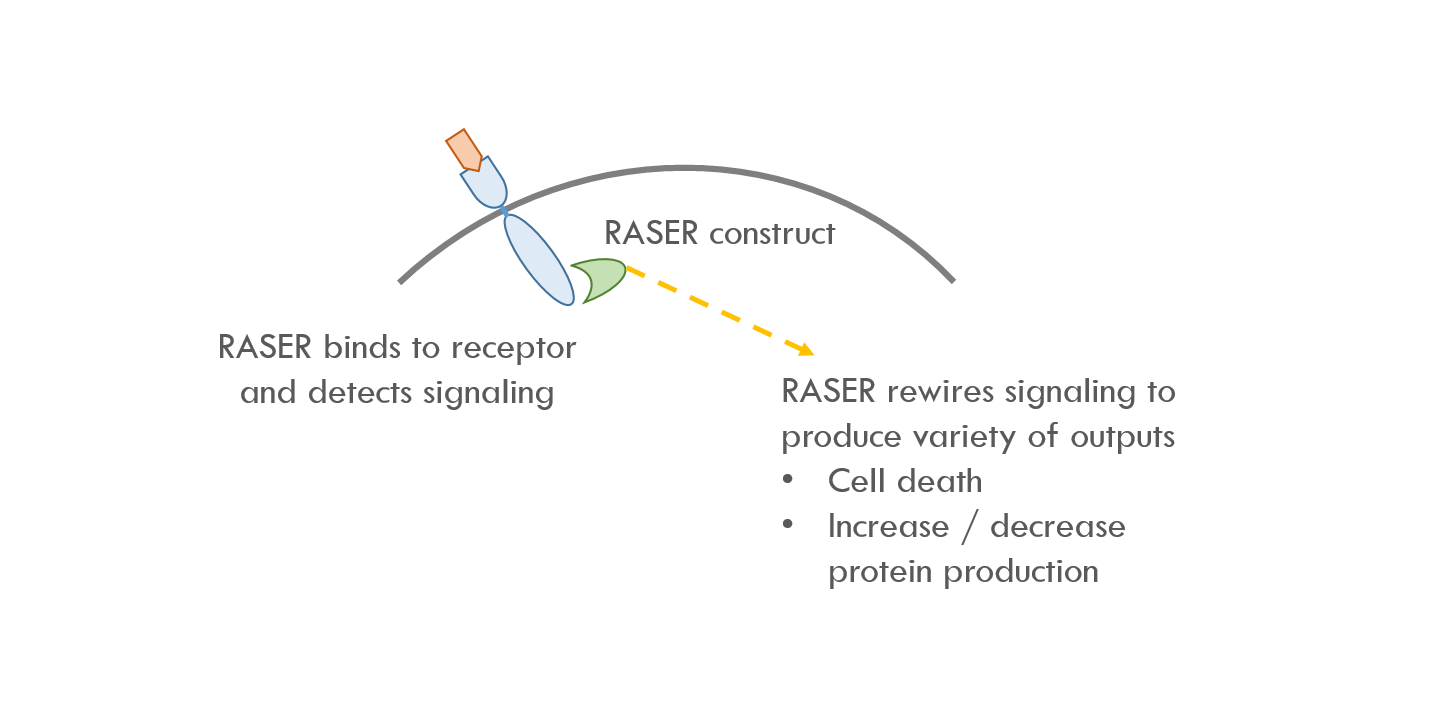
Designing RASER
Understanding the ErbB pathway
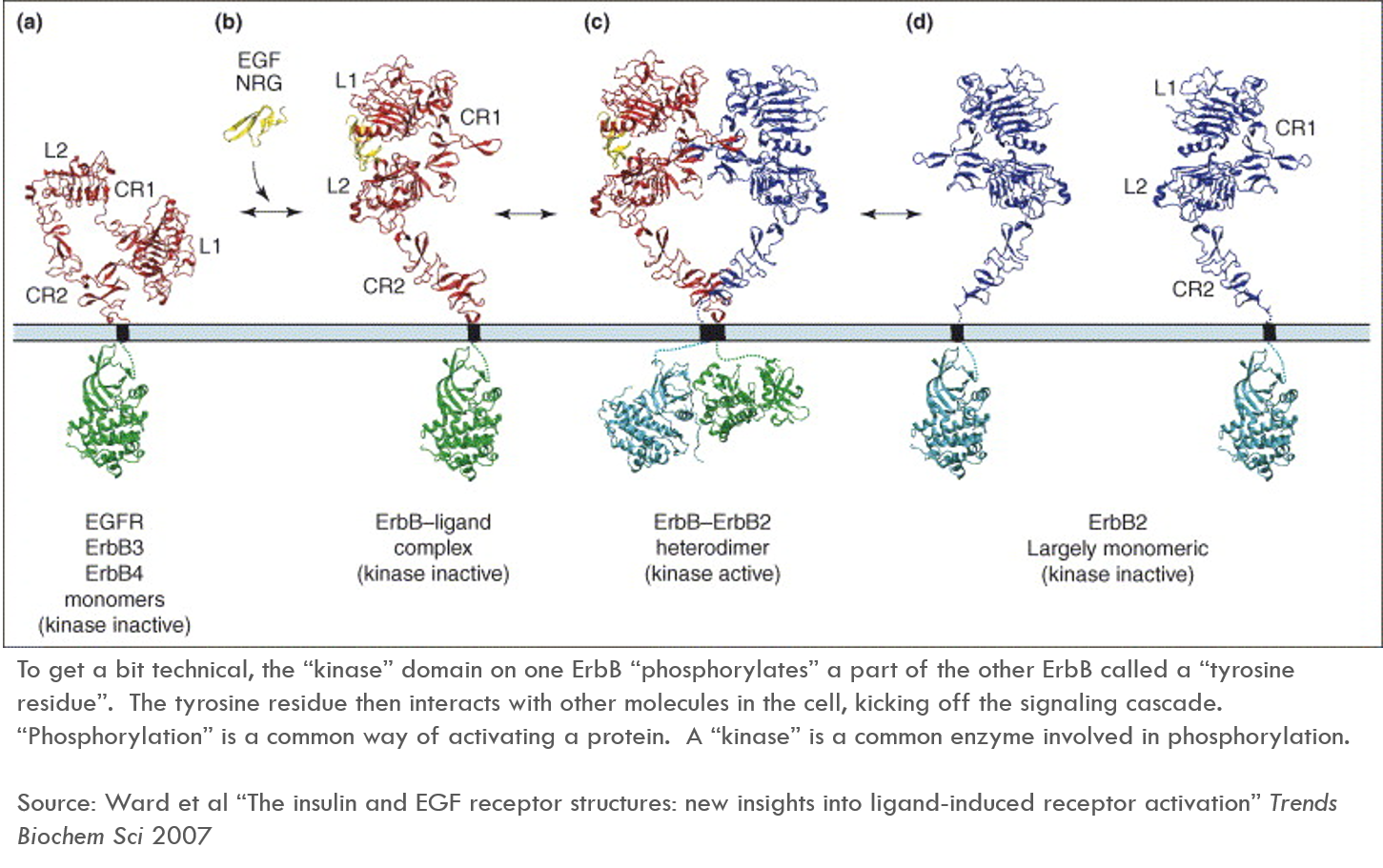
Understanding the paper requires some context on the ErbB pathway. ErbB, like most cell surface receptors, is a transmembrane protein – part of the protein extends out from the cell membrane and interacts with the extracellular environment, and part of the protein lies inside the cell (in the “cytoplasm”) and sends signals within the cell.
The below diagram describes how ErbB is activated. This diagram refers specifically to ErbB1, which is also known as EGFR. EGFR’s ligand (in yellow in the diagram) is EGF 4.
In healthy cells, ErbB1 is only active when it is bound by extracellular ligands. When ErbB1 binds its ligand, ErbB1 changes its shape, exposing a previously hidden region of the protein called CR1. CR1 sort of acts like an arm that reaches out and binds another ErbB receptor, bringing the two ErbB proteins together. This is called “dimerization” (a single protein is a monomer, a complex of two proteins is a dimer).
When two ErbB molecules are bound together, the intracellular part of one ErbB “activates” the intracellular part of another ErbB. These activated proteins then send signals throughout the cell, kicking off a “signaling cascade”.
Healthy vs. oncogenic signaling
In some cancers, ErbB is either mutated so that it is always active (instead of only being active in the presence of a specific ligand), or the cell just makes a lot more ErbB than usual. In either case, there is too much pro-growth signaling going on, which drives cancer.
But healthy cells also rely on ErbB signaling. To be therapeutically useful, RASER must be able to distinguish between healthy and oncogenic signaling. More on that later.
What is RASER?
RASER is a DNA cassette that codes for a set of synthetic proteins. When delivered into the nucleus via a vector (commonly a virus or a circular section of DNA called a plasmid), the cell’s natural genetic machinery will transcribe the DNA into RNA and then translate that RNA into protein. The proteins then move throughout the cell to execute the “program” of interest.
RASER contains two sets of synthetic proteins. I’ll call them “RASER protein A” and “RASER protein B” (they have more technical names in the paper). Their functions are described below:
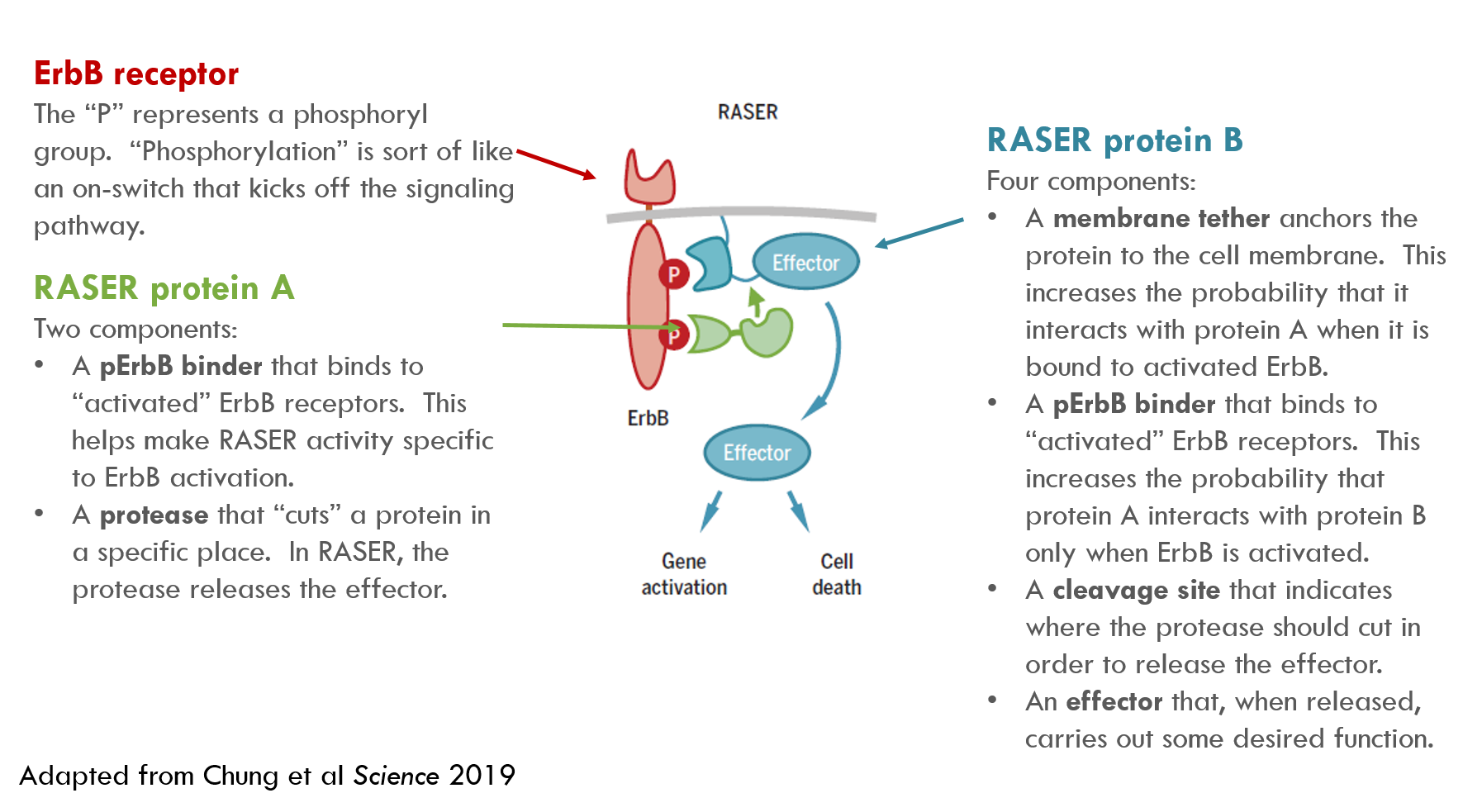
When protein A and protein B are in close proximity, a protease (protease = a protein that cuts other proteins) on protein A snips the effector from protein B, releasing the effector cargo to do its work.
Protein A and B can potentially interact even when they are not bound to activated ErbB. This is “off-target” activity, and we don’t want this – it could kill healthy cells. To minimize off-target activity, RASER includes a few components – specifically, the pErbB binders and the membrane tether – that increase the concentration of protein A and B at ErbB receptors, and thus increase the likelihood that they only interact when the receptor is active. The authors created a mathematical model that predicts how much off-target effector release will occur with different RASER designs.
Because ErbB signaling is important in healthy cells, RASER must be able to differentiate between healthy and oncogenic signaling. RASER accomplishes this by integrating signal over time: rather than sending a binary “kill or don’t kill” signal, RASER releases cargo that accumulates in the cell over time, and only kills a cell if a specific threshold is reached.
Creating, testing and optimizing RASER
The authors state two main design objectives for RASER:
- The system must be specific – it can differentiate normal signaling from oncogenic signaling
- The system must be programmable – it can convert the presence of an oncogenic signal to a variety of therapeutic outputs
Based on this specification, the authors designed an initial construct:
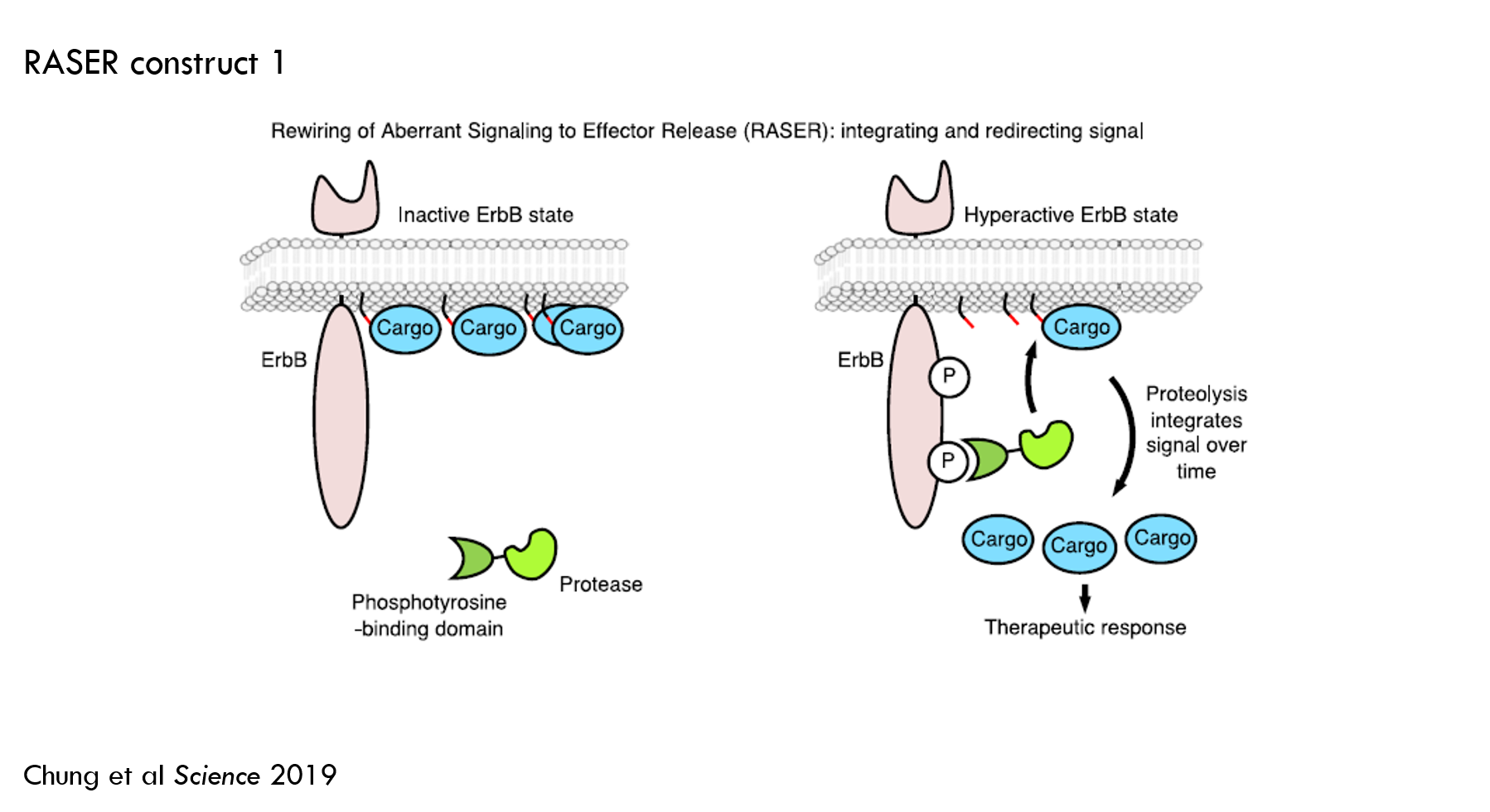
- Protein A (green in the above image): a protease fused to phosphotyrosine-binding domain (PTB).
- PTB binds to activated ErbB to detect the initiation of ErbB signaling
- To prevent unwanted cleavage of normal proteins, the authors used a viral protease 5. A viral protease won't cut any natural human proteins. Another benefit of this particular protease is that it can be blocked by an FDA-approved small molecule to temporarily turn RASER off.
- Protein B (blue oval with the red and black lines in the above image): effector fused to 1) a sequence that tells protein A’s protease where to cut and 2) a membrane tether.
- The cargo in this construct is orange fluorescent protein (OFP). OFP doesn’t change the cell’s function, but it is easy to measure, so it’s good for an initial test construct.
- The membrane tether increases the chances that protein B only interacts with protein A upon ErbB activation.
Testing RASER
This construct’s goal was to validate that their mathematical model accurately predicted the rate of effector release in ErbB “on” vs. “off” states. The authors used the following methodology:
- Measured the rate of orange fluorescent protein (OFP) release as a proxy for ErbB signaling
- Tested RASER in ErbB “on” cells and in ErbB “off” cells, and measured the difference in OFP levels
- The ErbB “on” arm was BT-474 cells. BT-474 is a human breast cancer cell line where ErbB2 (aka HER2) is overexpressed.
- The ErbB “off” arm used the same cell line, but they administered a small molecule, lapatinib, that blocks ErbB.
- Measured OFP levels using a technique called Western blot, a common molecular biology technique.
- Basically, they lysed cells (lysed = break down the cell membrane, making intracellular molecules accessible), collect the proteins, run the proteins through a gel that separates them by size, transfer the proteins to a membrane, then label the proteins for detection. Proteins are generally labeled with two antibodies: a primary antibody that is specific for the protein of interest and a secondary antibody that binds to the primary antibody. The secondary antibody is labeled – often with a fluorescent molecule – so it can be detected by imaging or visual inspection.
- Tested four RASER constructs made of combinations of two different proteases and two different substrates sequences:
- NS3 protease with a cofactor (medium speed of cleaving) and without a cofactor (slow speed of cleaving)
- EDVVCC substrate (higher affinity 6 for the protease) and DEMEEC (medium affinity)
- After 24 hours they measured the level of various proteins:
- Uncleaved substrate
- Released OFP
- GAPDH (an internal control)
I don’t have enough knowledge to critique the study design, but it makes sense to me as an initial proof of concept. I think it also might make sense to measure a marker of ErbB signaling to verify that lapatinib is blocking ErbB signaling. This would also show whether RASER blocks ErbB signaling – if RASER did not block ErbB signaling, it would be much more useful as a potential therapy. In a separate experiment (in Fig 3C and 3D) the authors observed that RASER reduced, but did not completely block, ErbB signaling as measured by pAkt and pErk. Supplemental Figure 8B shows that RASER does not block ErbB signaling in SK-BR-3 cells but does block ErbB signaling in LN-229:EGFRvIII cells. It would be nice to better understand this.
It’s easy to imagine more ways to play around with this – testing different proteases, substrate sequences, ErbB binders, etc. They tested a few of these (protease and substrate sequence) in the paper. Changing the protease and substrate sequence affects the total amount of protein cleavage in both ErbB “on” and “off” states. Faster-cleaving proteases and higher affinity substrates result in more substrate cleavage.
Preliminary results
Here’s an image of the Western blot (Fig 1F) and a chart quantifying substrate cleavage (Fig 1G). Darker blobs indicate more protein. Larger proteins are closer to the top. GAPDH is an internal control to make sure each “column” has the same number of cells. PTB-pro is ErbB-binder + medium-speed protease, PTB-pro(54A) is ErbB-binder + the slow protease. EDVVCC is higher affinity substrate sequence, DEMEEC is medium affinity. CAAX is the membrane tether.
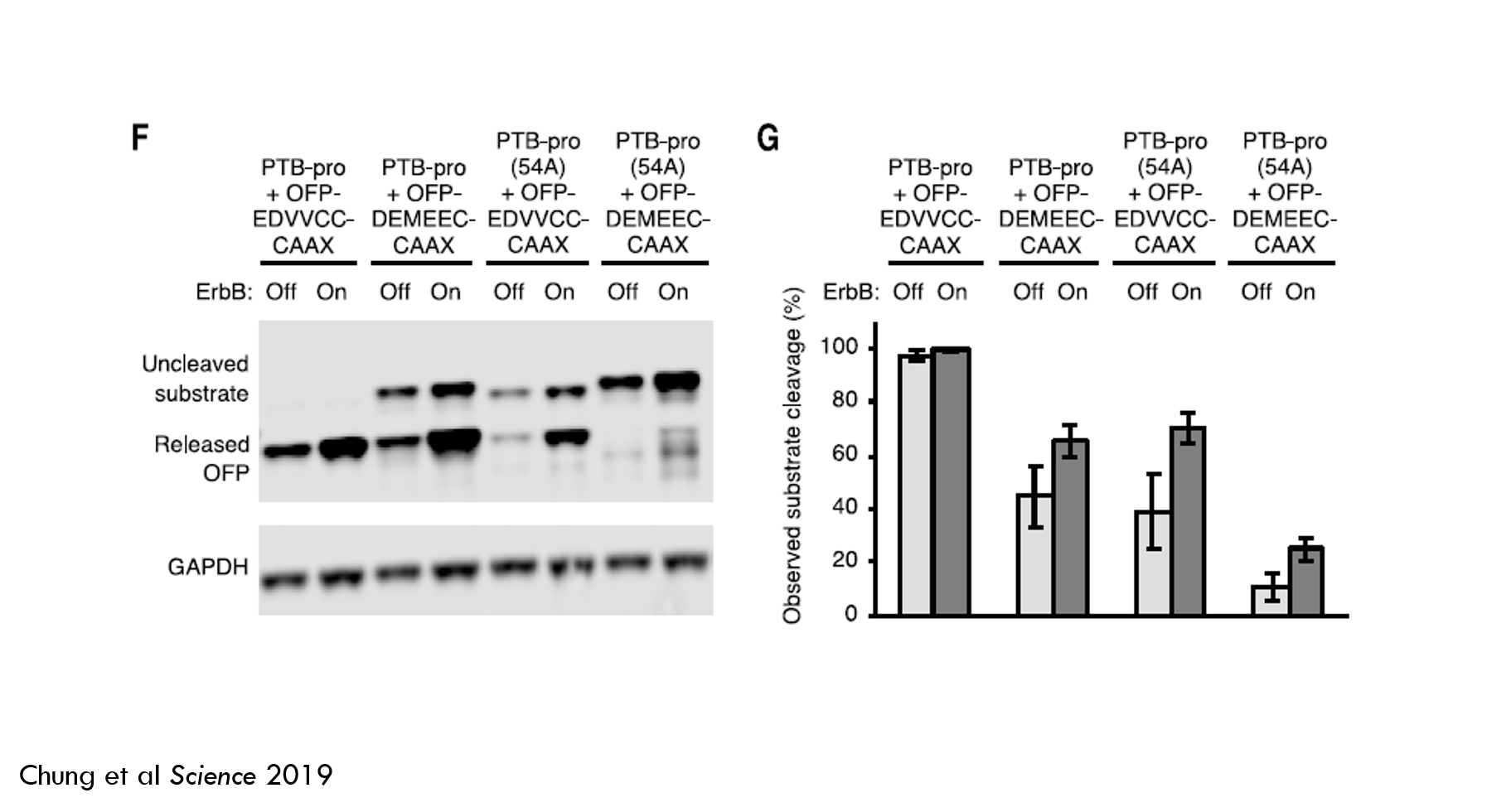
The faster protease + high-affinity substrate cleaved almost 100% of the effector cargo in both ErbB-off and ErbB-on states. This is not a very good detector of oncogenic ErbB signaling. The other arms did better, but still weren’t great. So there’s more work to do.
They did show that the mathematical model predicted activity fairly well, so they could use this model to engineer subsequent designs.
Testing RASER
The authors then tweaked several aspects of RASER to improve its performance. First, they added an ErbB binder called SH2 to protein B. In the prior construct, protein B had a membrane tether, but it didn’t have an ErbB binder. The authors predicted that this ErbB binder would reduce “off-target” effector release.
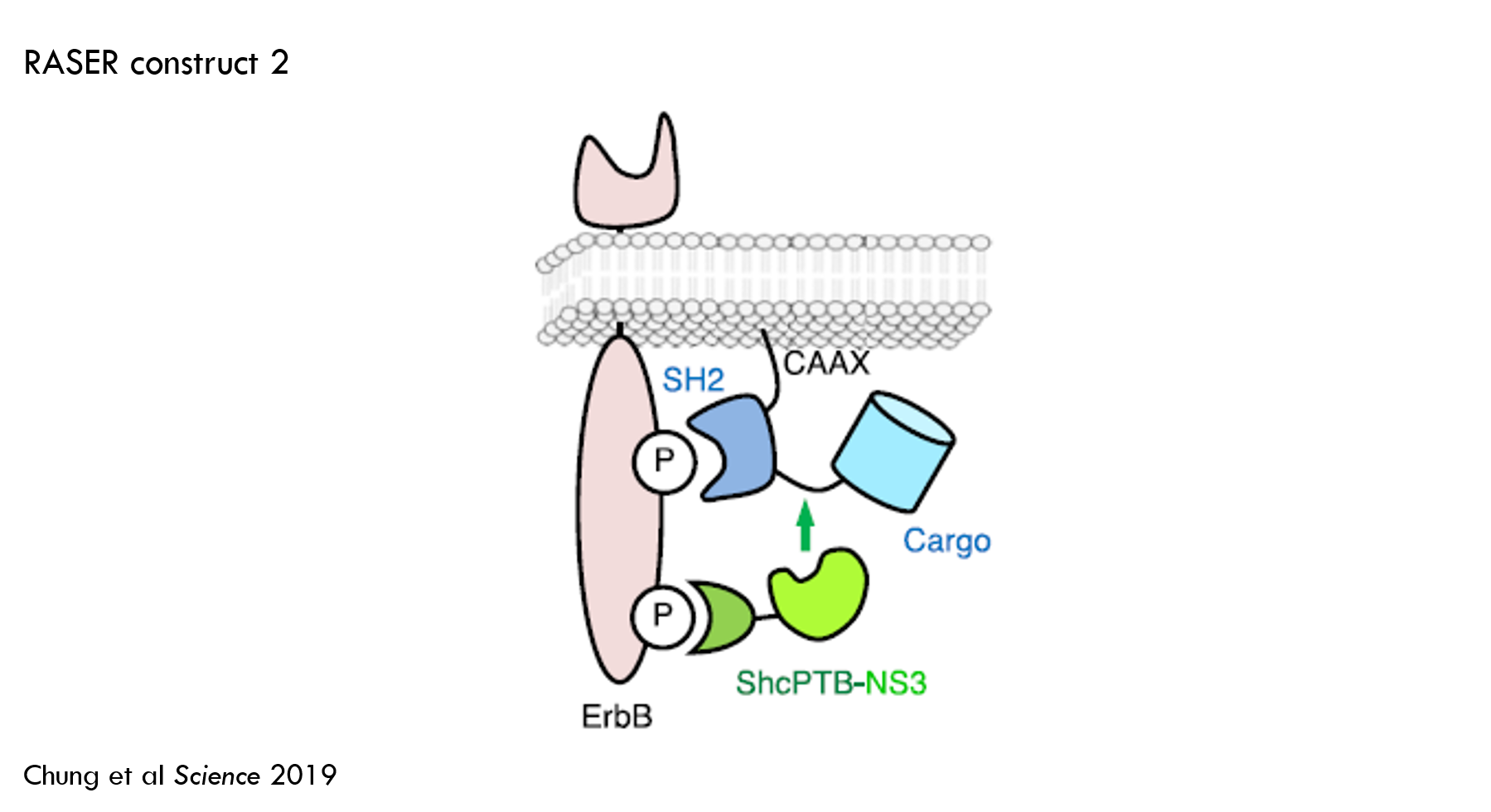
This improved the ability to detect oncogenic vs. healthy ErbB signaling, but there was still significant off-target cleavage (30% of substrate was cleaved in ErbB “off” state).
To reduce off-target effector release, they engineered protein A to degrade unless it was bound to ErbB. They accomplished this by adding a “degron” to protein A. A degron increases the rate of protein degradation by recruiting a protein called ubiquitin ligase, which tags the protein for degradation by the cell's "proteasome". When protein A is bound to ErbB, the degron is hidden, so the protein can’t be tagged for degradation.
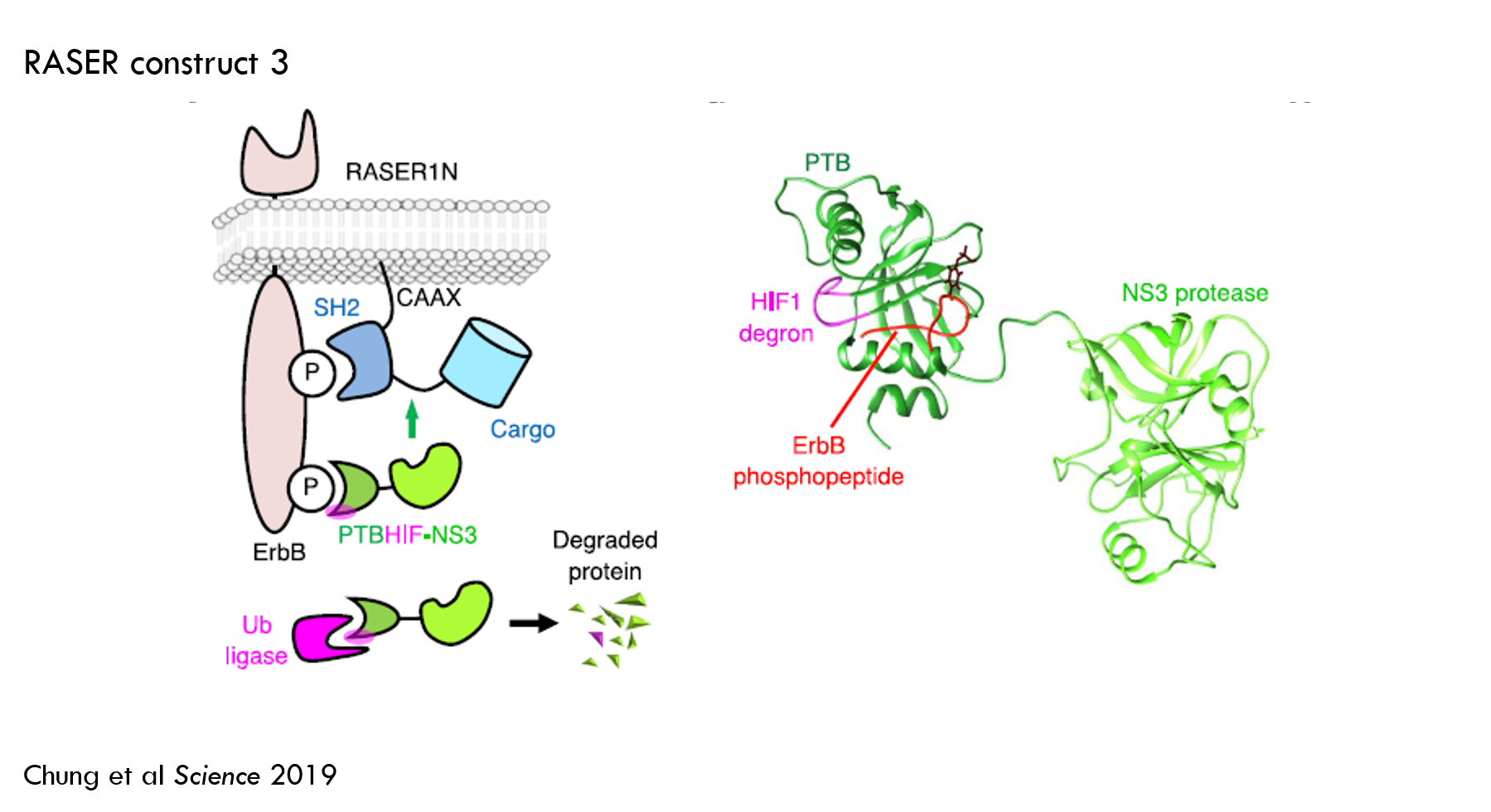
This reduced off-target effector release to about 10% and increased “on-target” release to about 80%. This worked well enough for the purposes of the paper, so the authors selected this design for subsequent work.
As an aside, I thought this was the coolest part of the paper. The fact that they can think of this and actually make it work sort of blows my mind. They had an idea, added some DNA to their sequence, and all of a sudden they can dynamically control the expression of a protein in the cell. Protein engineering is amazing.
Exploring RASER’s versatility
Next, the authors explored the generalizability, specificity and reproducibility of RASER. Specifically, they showed that:
- RASER can be designed to release cargo from both sides of protein B.
- RASER can differentiate between healthy and oncogenic signaling in different cancer cell lines.
- RASER can detect both ErbB1 (aka EGFR) signaling and ErbB2 (aka HER2) signaling.
- RASER does not respond to high ligand-induced stimulation of ErbB-normal cells, but it does respond to normal ligand-induced stimulation of ErbB-overexpressing cancer cell lines.
- ErbB activity is more clearly detectable by measuring RASER output than it is by measuring endogenous Akt and Erk phosphorylation.
This is some nice preliminary data suggesting the generalizability, reproducibility and selectivity of RASER and sets the stage for a lot of interesting follow-up work.
Testing therapeutic effector cargos
In the previous experiments, the cargo was OFP. This is fine for developing the system, but it isn’t particularly exciting from a therapeutic perspective. Fluorescent protein won’t cure cancer.
The first therapeutic effector they tested was a cell-killing protein. They screened a group of apoptosis-inducing molecules (apoptosis is a form of programmed cell death) and selected one called Bid (Fig S9A of the paper shows the screening experiment).
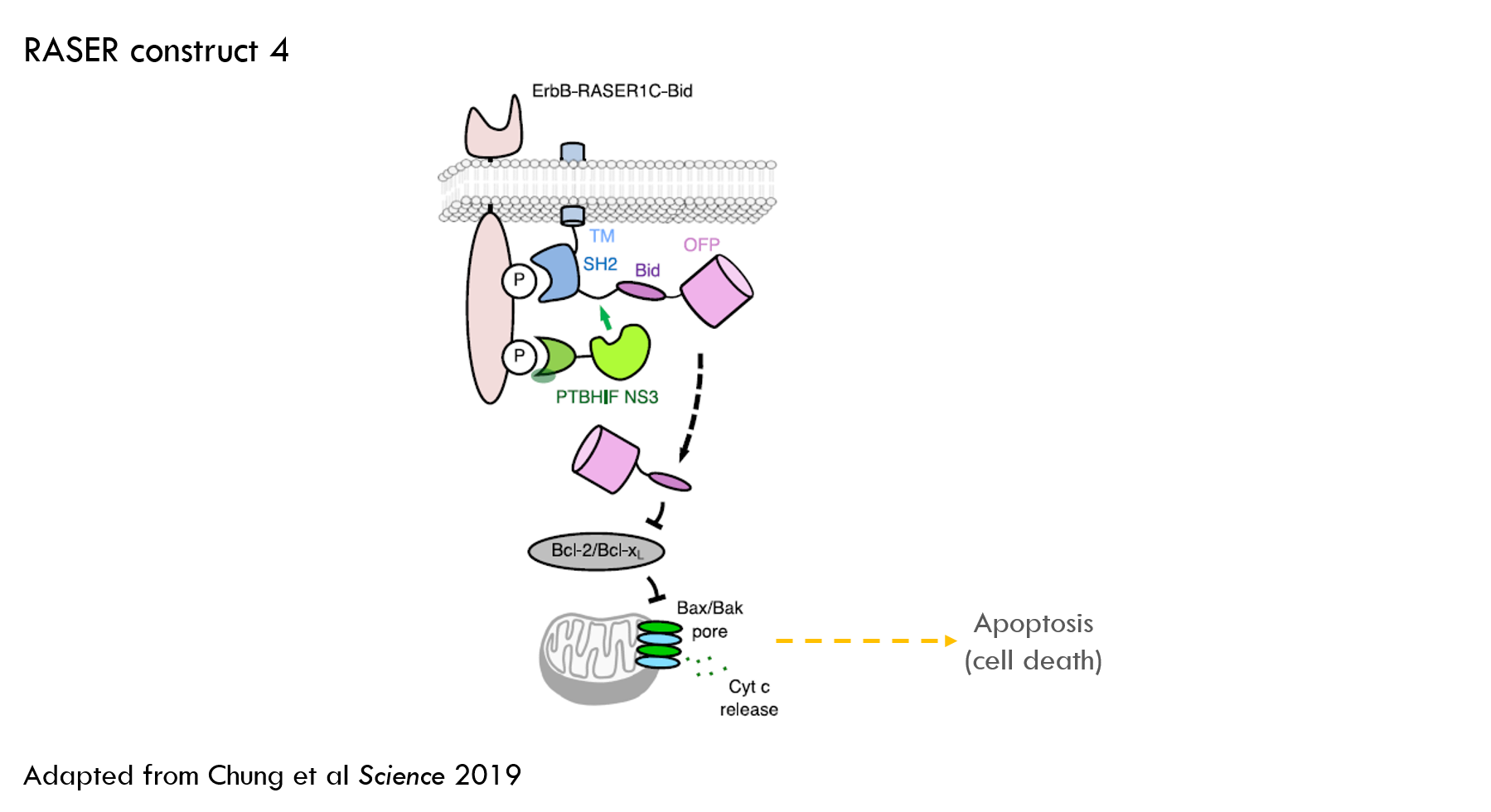
Testing cancer cell killing
The authors used a similar experimental setup to the previous experiments, but additionally measured PARP cleavage as a marker for apoptosis. They delivered Bid alone as a control.
RASER increased levels of PARP cleavage (which is a marker for cell death) to 99% of the level of Bid alone. This activity was blocked by lapatinib, the ErbB inhibitor. This was a nice result – RASER is as effective at killing cells as administering a cell-killing protein, but only in ErbB “on” cells.
The authors ran the same experiment in MCF-7 cells. MCF-7 is an “ErbB normal” cancer cell line. We wouldn’t want RASER to kill these cells, but we would expect Bid alone to kill the cells. This is in fact what they saw.
The authors also tested this RASER construct in several other cancer cell lines with oncogenic ErbB and saw similar levels of cancer cell killing, suggesting the tool is somewhat robust across different cell models.
In previous experiments, they delivered RASER with a plasmid, but they also tested viral delivery with lentivirus. Viral delivery was effective at getting RASER into cells. This is important because to get DNA into human cells you typically need a viral vector (you can get DNA into cells in vitro with various other methods).
Increasing protein expression with RASER
They also tested a “transcriptional activator” as the effector to see if RASER could increase expression of specific proteins. Transcription is the first step of expressing proteins from DNA, so activating transcription is a way to increase protein expression.
They first tested a synthetic transcription factor, tTA, and showed that they were able to increase production of a fluorescent protein that is expressed with the help of tTA. Then they tested an endogenous (ie a natural as opposed to synthetic) transcription factor, which also worked.
Then they used a custom CRISPR-Cas9 domain as cargo. This was the second coolest part of the paper for me. These cargoes can be used to regulate expression of almost any endogenous gene.
For those not familiar, CRISPR-Cas9 is a construct made of a guide RNA (gRNA) and a protein called Cas9. Cas9 is an endonuclease – a protein that cuts DNA. gRNA guides Cas9 to a specific gene, and Cas9 cuts that gene.
CRISPR-dCas9 is a clever play on the “gene cutting” version of CRISPR-Cas9. Instead of using normal Cas9, which cuts DNA, they use a “catalytically dead” version (dCas9 for short). dCas9 is unable to cut DNA; it just sits there, bound to the DNA. This temporarily blocks gene expression by preventing other proteins from binding to DNA and initiating transcription.
Researchers have found a way to tweak this approach to increase rather than block protein expression. They fuse a “transcriptional activation domain” to dCas9 that recruits other proteins to transcribe the DNA. I don’t know the details of how this works, but you can theoretically use one customizable tool to increase or decrease expression of a variety of genes.
They were able to use dCas9 cargo to promote expression of a reporter gene and therapeutic gene, GM-CSF. GM-CSF stimulates the immune system to attack cells expressing specific molecules, so in this case can “turbo-charge” the immune response to cancer.
Finally, they tested RASER’s cancer cell killing ability vs. some approved therapeutic agents in a few in vitro cancer models. This is nice data, but additional models would be more informative of RASER’s therapeutic potential.
Next steps
There’s a lot you could do with RASER. I’ll briefly discuss some of my own personal considerations for how RASER might be developed as a therapeutic tool.
In my mind, there are three main areas to address:
- Assessment of RASER as a therapy for HER2+ breast cancer and / or EGFR-mutant lung cancer
- Assessment of the broader potential of RASER as a therapeutic platform
- Confirmatory assessment of the robustness of the science
The main cell line they studied was BT-474, which is a HER2+ breast cancer cell line, so it makes sense to think about RASER as a potential treatment for HER2+ breast cancer.
HER2+ breast cancer is a very large market. The first-line standard of care is generally Herceptin (aka trastuzumab) + taxane for Stage 1 cancer, then Herceptin + Perjeta for later-stage cancer. Standard of care works works well in Stage 1, with seven-year disease free survival rates of 93% but outcomes are worse in later-stage cancers, with 3-year overall survival rates in metastatic cancer of about 70%. Herceptin did $3B in US sales and almost $7B in worldwide sales in 2018.
HER2+ breast cancer is a competitive market, but there is room for new therapies. 15% of breast cancer patients are HER2+ and candidates for Herceptin, but 45% of patients have low levels of HER2. These patients are not considered HER2 positive and don’t respond to Herceptin. It seems there is some unmet need in HER2+ metastastic patients who don’t respond to Herceptin + Perjeta, but the larger market is probably in low-HER2 patients.
Daiichi Sankyo has an antibody-drug conjugate (trastuzumab fused to a chemotherapy) that is being tested in low-HER2 patients. The trastuzumab component of Daiichi’s drug binds to HER2, and then the cell internalizes the drug. Once in the cell, the chemo is released. The drug was valued at almost $7B in a recent deal with AstraZeneca .
There appears to be room for better treatments for breast cancer. But how likely is RASER to be a best-in-class treatment?
In the paper, they tested RASER against approved breast cancer drugs to see if it worked as well as standard of care. However, it isn’t clear whether the comparators they used (paclitaxel and lapatinib) were representative of standard of care. Ultimately, it would depend on the specific indication being pursued, but it would be nice to see some data against used trastuzumab + taxane or Perjeta (paclitaxel, which the authors used as a comparator, is a taxane molecule, but it is more appropriate to use paclitaxel + trastuzumab as the comparator).
It is also important to test RASER in gold-standard cancer models. I don’t know much about the specific in vitro models they used, but obviously they’d need to test the drug in a gold-standard animal model, potentially a patient-derived xenograft mouse model.
Testing an experimental drug in gold-standard animal models against the standard of care is a normal step for all projects. There are a few additional challenges specific to RASER. RASER is a very novel therapeutic modality -- there are no drugs that work like RASER available today. So it has some unique considerations in addition to its advantages.
A major challenge is delivery. Many viral vectors can generally only be dosed systemically once. After one dose, the body develops immunity to the vector and rejects subsequent doses. So you’d have to get the first dose exactly right. This has proven to be a major challenge with other gene therapies. Some viruses have decent ability to penetrate tumor tissues, which could be an advantage, but intratumoral injection may not be able to clear all metastases. Finding the right vector and route of administration is critical.
It is particularly difficult because you’d presumably need at least one virus to infect each cancer cell if you want to kill the whole tumor. To do this, you’d probably need to dose a very high number of viruses (and if you don’t get enough the first time, you may not get another shot). There have been some reports that too much virus can damage the liver.
To reduce the dose required for meaningful tumor killing, it may be possible to engineer RASER to stimulate a stronger immune response to the tumor (perhaps by adding GM-CSF to RASER, as they did in the paper), but there is no evidence yet that RASER could cause a systemic anti-tumor response.
Perhaps more concerning is the risk that a high dose kills a lot of healthy cells. HER2 is widely expressed and important in healthy breast cell function. The paper showed that the RASER-Bid (apoptotic cargo) still killed some ErbB-negative cells (Figure 4C and 4D). The data in Figure 6 look better, but this is still an important risk, especially if high doses are required.
Another risk is that RASER blocks healthy ErbB signaling. The data presented in the paper seemed mixed in this regard: Fig 3D showed that RASER reduced, but didn’t fully block, ErbB signaling, while Fig S8B showed RASER did not block ErbB signaling in SK-BR-3 cells but did block ErbB signaling in LN-229-EGFRvIII cells.
Finally, independently confirming the robustness of the science is a standard step in bringing something out of academia (regardless of who the inventor is and what journal it was published in). You can do this by reading the paper critically, talking to the researchers, and discussing the work with technical experts. Ultimately you’ll often re-do key experiments, especially if the work is novel and hasn’t yet been reproduced.
Other applications
The HER2+ breast cancer indication seems worth further exploration, but there are other targets that might be even more compelling. KRAS is an obvious one. Kras is one of the most common oncogenes, but it is virtually impossible to drug. It is an intracellular protein, so can’t be drugged with antibodies (antibodies are unable to get inside of cells). It is a “smooth” protein with no spots for small molecules to latch onto, so small molecules can’t hit it.
RASER could theoretically kill Kras-driven cancer cells. I’m not sure what they’d use as the KRAS-targeting component for a KRAS-RASER, or how they’d differentiate between healthy and cancer cells, but it seems like a logical thing to test. It appears the lab is in fact working on this (per a Biocentury Innovations article from May 23, 2019).
Beyond ErbB, KRAS and cancer, there are a multitude of potentially interesting applications. Figuring out which are the most promising is beyond the scope of this post, but the breadth of potential applications may be the most exciting thing about RASER.
You may also like...
Valuing drugs and biotech companies
Valuations of biotech startups from Series A to IPO
List of recently funded biotech startups
Bay Bridge Bio Startup Database
The world's most expensive drug? A case study of Zolgensma
Did you enjoy this article?
Then consider joining our mailing list. I periodically publish data-driven articles on the biotech startup and VC world.
1 This post will focus on comprehension. It is also important to review the methods and materials critically, but that is beyond the scope of this post. When it comes to interpreting scientific publications (even in top journals), trust but verify!
2 Receptors can also be located inside of the cell, but for this post I’ll use “receptor” to mean “cell surface receptor”.
3 Two very common forms of cancer are driven by overactive ErbB: EGFR-mutated lung cancer (EGFR is also known as ErbB1) and HER2+ breast cancer (HER2 = ErbB2).
4 EGF = epidermal growth factor. EGFR = epidermal growth factor receptor.
5 The authors used a mathematical model to pick which protease to use. They selected a protease from Hepatitis C virus called NS3 because it was predicted to release less effector cargo at baseline than the other proteases they tested. They tested two types of NS3, one that does not require a cofactor and one that does. A cofactor is a molecule that is required for an enzyme (like a protease) to work.
6 Affinity is the strength of binding between a protein and its ligand.